Nhà sản xuất
Novartis Pharma
Thành phần
Mỗi viên: Ceritinib 150mg.
Mô tả
Viên nang cỡ #00 màu trắng đục và xanh đục, nắp màu xanh đục có dòng chữ bằng mực đen “LDK 150MG”, thân màu trắng đục có dòng chữ bằng mực đen “NVR”, chứa bột màu trắng đến gần trắng.
Dược lý
Nhóm dược lý: Tác nhân chống ung thư và miễn dịch.
Mã ATC: L01XE28.
Cơ chế tác động
Ceritinib là một thuốc dùng đường uống ức chế mạnh và rất chọn lọc trên ALK kinase. Ceritinib ức chế quá trình phosphoryl hóa tự động của ALK, quá trình phosphoryl hóa qua trung gian ALK các protein truyền tín hiệu xuôi dòng và quá trình tăng sinh của các tế bào ung thư phụ thuộc ALK cả in vitro và in vivo.
Sự chuyển vị ALK quyết định biểu hiện của protein tổ hợp và dẫn đến các bất thường trong tín hiệu ALK trong ung thư phổi không tế bào nhỏ. Trong phần lớn trường hợp ung thư phổi không tế bào nhỏ, EML4 là một phần chuyển vị của ALK sản sinh ra các protein tổ hợp EML4-ALK có chứa các vùng protein kinase của ALK liên kết với thành phần nitơ tận cùng của EML4. Ceritinib đã được chứng minh có tác dụng ức chế hoạt tính của EML4-ALK trên các dòng tế bào ung thư phổi tế bào không nhỏ (H2228), dẫn đến ức chế quá trình tăng sinh của tế bào in vitro và sự thoái triển của khối u ghép H2228 trên chuột nhắt và chuột cống.
ĐẶC TÍNH DƯỢC LỰC HỌC
Tác dụng ức chế hoạt tính ALK kinase và các con đường truyền tín hiệu qua trung gian ALK trên Karpas 299 (dòng tế bào lympho) và trên H2228 (dòng tế bào ung thư phổi) của ceritinib đã được chứng minh phụ thuộc vào liều. Tác dụng ức chế của ceritinib dẫn đến ức chế tăng sinh của tế bào ung thư in vitro và sự tiến triển của khối u in vivo trên các mô hình ghép khối u ở chuột nhắt và chuột cống. Tác dụng ức chế enzym của ceritinib gấp khoảng 20 lần so với crizotinib trong các thử nghiệm ức chế ALK kinase (IC50 cho tác dụng ức chế ALK của ceritinib là 0,15 nM và của crizotinib là 3 nM). Trong một bộ kinase gồm 36 enzym, ceritinib chỉ ức chế 2 kinase khác với hoạt lực yếu hơn khoảng 50 lần so với tác dụng ức chế ALK. Tất cả các kinase khác trong bộ các enzym này đều bị ức chế kém hơn trên 500 lần so với tác dụng ức chế ALK cho thấy mức độ chọn lọc cao của thuốc trên enzym ALK. Một nghiên cứu dược lực học đơn liều và nghiên cứu hiệu quả của liều lặp lại được thực hiện trên mô hình u lympho Karpas 299 và ung thư phổi H2228 cho thấy để giảm được sự tiến triển của khối u cần giảm 60% đến 80% con đường truyền tín hiệu ALK.
CÁC NGHIÊN CỨU LÂM SÀNG
Ung thư phổi không tế bào nhỏ (NSCLC) tiến xa tại chỗ hoặc di căn, dương tính với ALK, chưa được điều trị trước đây - Nghiên cứu A2301 ngẫu nhiên pha 3 (ASCEND-4)
Hiệu quả và độ an toàn của Spexib trong điều trị bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC) dương tính với ALK, tiến xa tại chỗ hoặc di căn, có và không có di căn não, chưa được điều trị toàn thân bằng liệu pháp chống ung thư trước đây (bao gồm thuốc ức chế ALK) ngoại trừ liệu pháp bổ trợ hoặc tân bổ trợ, đã được chứng minh trong nghiên cứu A2301 pha 3, đa trung tâm toàn cầu, ngẫu nhiên, nhãn mở.
Tiêu chí hiệu quả chính là thời gian sống còn không tiến triển bệnh (PFS), như được xác định bởi Ủy ban xem xét độc lập, mù với phân bổ điều trị (BIRC), theo tiêu chuẩn đánh giá đáp ứng về khối u đặc (RECIST) 1.1. Tiêu chí phụ quan trọng là thời gian sống còn toàn bộ (OS). Các tiêu chí phụ khác bao gồm tỷ lệ đáp ứng toàn bộ (ORR), thời gian đáp ứng (DOR), tỷ lệ kiểm soát bệnh (DCR) và thời gian đến khi đáp ứng (TTR) được xác định bởi BIRC và nghiên cứu viên và kết quả do bệnh nhân báo cáo (PRO), bao gồm các triệu chứng liên quan đến bệnh, chức năng và chất lượng cuộc sống liên quan đến sức khỏe.
Tỷ lệ đáp ứng nội sọ toàn bộ (OIRR), tỷ lệ kiểm soát bệnh nội sọ (IDCR) và thời gian đến khi đáp ứng nội sọ (DOIR) được xác định bởi bác sĩ chuyên khoa X-quang thần kinh của BIRC theo RECIST 1.1 sửa đổi (tức là lên đến 5 tổn thương trong não) đã được sử dụng để đánh giá hoạt tính chống khối u trong não.
Các bệnh nhân được phép tiếp tục điều trị nghiên cứu được phân bổ sau tiến triển ban đầu trong trường hợp tiếp tục có lợi ích lâm sàng theo ý kiến của nghiên cứu viên. Các bệnh nhân được phân ngẫu nhiên vào nhóm hóa trị liệu có thể bắt chéo để dùng ceritinib dựa trên sự tiến triển bệnh được xác định bởi BIRC theo RECIST.
Tổng cộng có 376 bệnh nhân được chọn ngẫu nhiên theo tỷ lệ 1:1 (phân tầng theo tình trạng hoạt động cơ thể theo Tổ chức Y tế thế giới (WHO), hóa trị liệu bổ trợ/tân bổ trợ trước đó và có/không có di căn não lúc sàng lọc) để dùng ceritinib (750 mg/ngày, lúc đói) hoặc hóa trị liệu (dựa trên lựa chọn của nghiên cứu viên - pemetrexed (500 mg/m2) cộng với cisplatin (75 mg/m2) hoặc carboplatin (diện tích dưới đường cong (AUC) 5-6), dùng mỗi 21 ngày). Những bệnh nhân đã hoàn thành 4 chu kỳ hóa trị liệu (dẫn nhập) mà không có bệnh tiến triển sau đó được dùng pemetrexed (500 mg/m2) điều trị duy trì đơn độc mỗi 21 ngày. Một trăm tám mươi chín (189) bệnh nhân được chọn ngẫu nhiên để dùng ceritinib và một trăm tám mươi bảy (187) bệnh nhân được chọn ngẫu nhiên để dùng hóa trị liệu.
Độ tuổi trung vị tổng thể là 54 tuổi (từ 22-81 tuổi); 78,5% bệnh nhân trẻ hơn 65 tuổi. Tổng cộng có 57,4% bệnh nhân là nữ. 53,7% nhóm đối tượng nghiên cứu là người da trắng, 42,0% là người châu Á, 1,6% là người da đen và 2,6% là các chủng tộc khác. Đa số bệnh nhân bị ung thư biểu mô tuyến (96,5%) và chưa bao giờ hút thuốc lá hoặc đã từng hút thuốc lá trước đây (92,0%). Tình trạng hoạt động cơ thể theo thang điểm của Nhóm hợp tác ung thư miền đông (ECOG) theo thứ tự là 0/1/2 ở 37,0%/56,4%/6,4% bệnh nhân, và 32,2% có di căn não thần kinh ổn định (có triệu chứng hay không) lúc ban đầu. Đặc điểm bệnh ban đầu rất đồng đều giữa hai nhóm điều trị.
Thời gian theo dõi trung vị là 19,7 tháng (từ ngày chọn ngẫu nhiên đến ngày khóa dữ liệu).
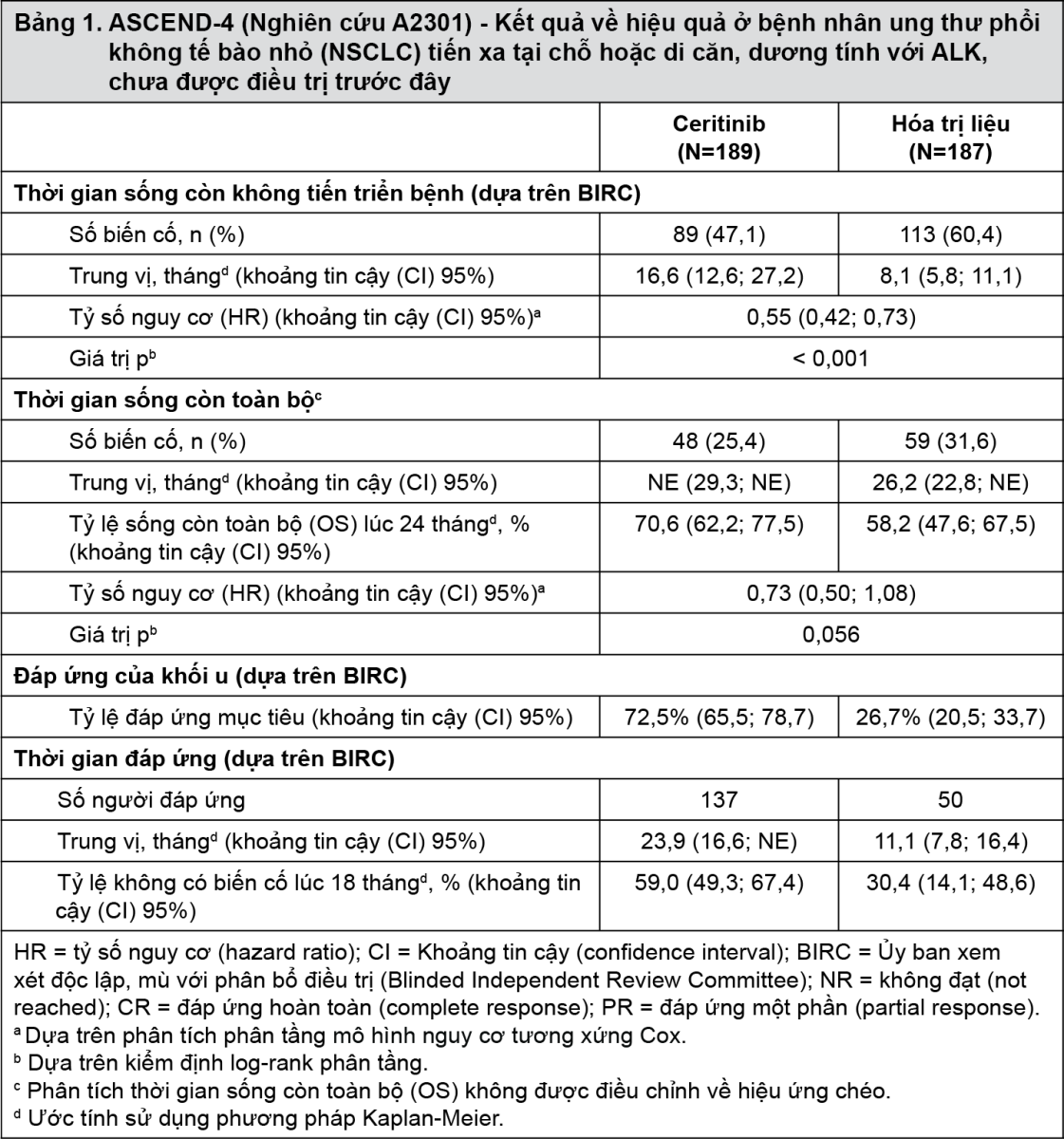
Nghiên cứu này đã đáp ứng mục tiêu chính cho thấy sự cải thiện có ý nghĩa thống kê và có ý nghĩa lâm sàng về thời gian sống còn không tiến triển bệnh (PFS) theo đánh giá của BIRC với sự giảm nguy cơ ước tính là 45% ở nhóm ceritinib so với nhóm hóa trị liệu (Tỷ số nguy cơ (HR): 0,55 với khoảng tin cậy (CI) 95%: 0,42; 0,73; p<0,001). Thời gian sống còn không tiến triển bệnh (PFS) trung vị là 16,6 tháng (khoảng tin cậy (CI) 95%: 12,6; 27,2) đối với nhóm ceritinib và là 8,1 tháng (khoảng tin cậy (CI) 95%: 5,8; 11,1) đối với nhóm hóa trị liệu (xem Bảng 1 và Hình 1).
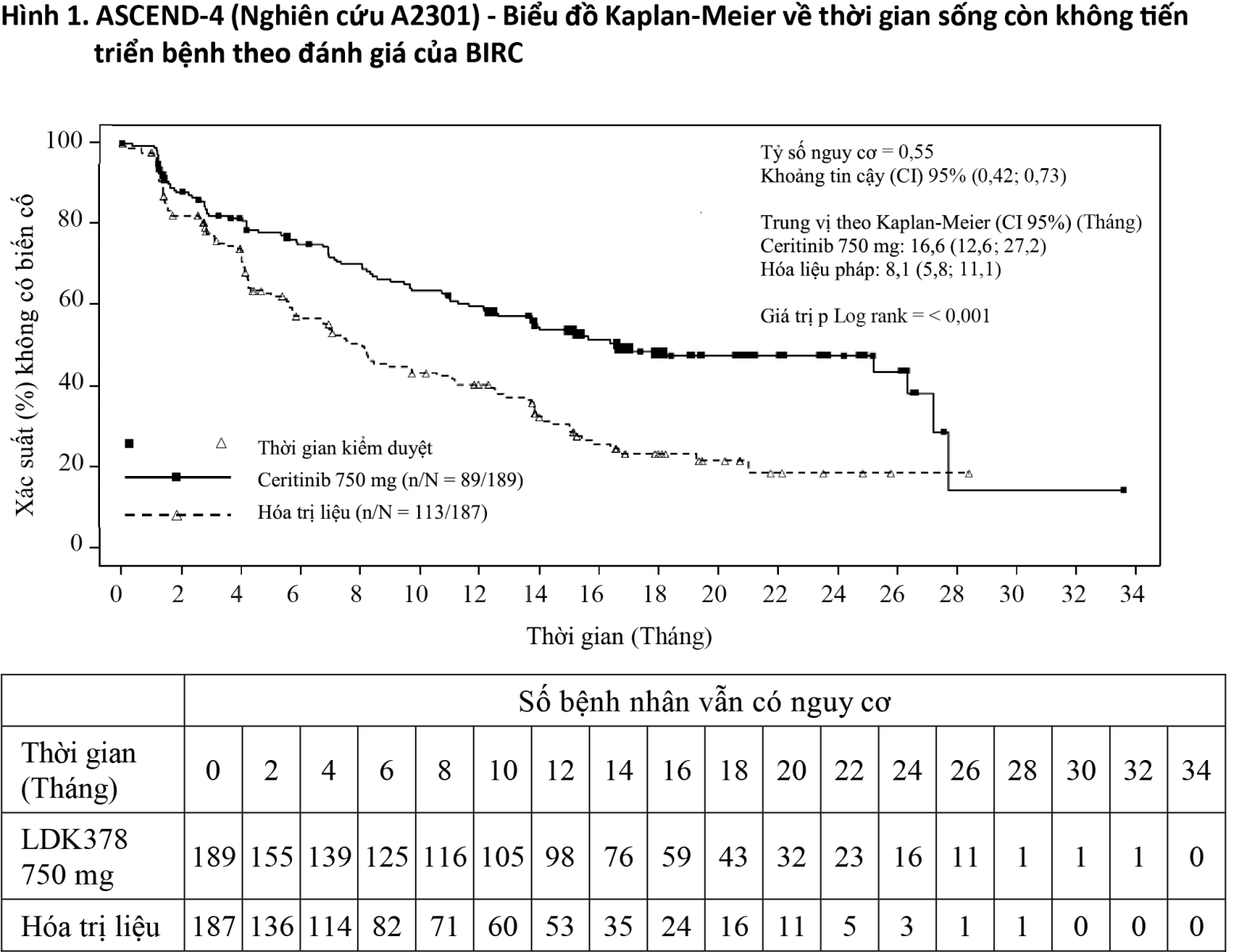
Lợi ích của ceritinib về thời gian sống còn không tiến triển bệnh (PFS) vượt trội so với hóa trị liệu là rõ ràng và đồng nhất theo đánh giá của nghiên cứu viên và ở các phân nhóm khác nhau bao gồm tuổi, giới tính, chủng tộc, nhóm hút thuốc lá, tình trạng hoạt động cơ thể theo thang điểm của Nhóm hợp tác ung thư miền đông (ECOG) và gánh nặng bệnh tật (xem Hình 2).
Ceritinib cũng cải thiện có ý nghĩa tỷ lệ đáp ứng toàn bộ (ORR) theo đánh giá của BIRC so với hóa trị liệu với đáp ứng lâu bền (xem Bảng 1).
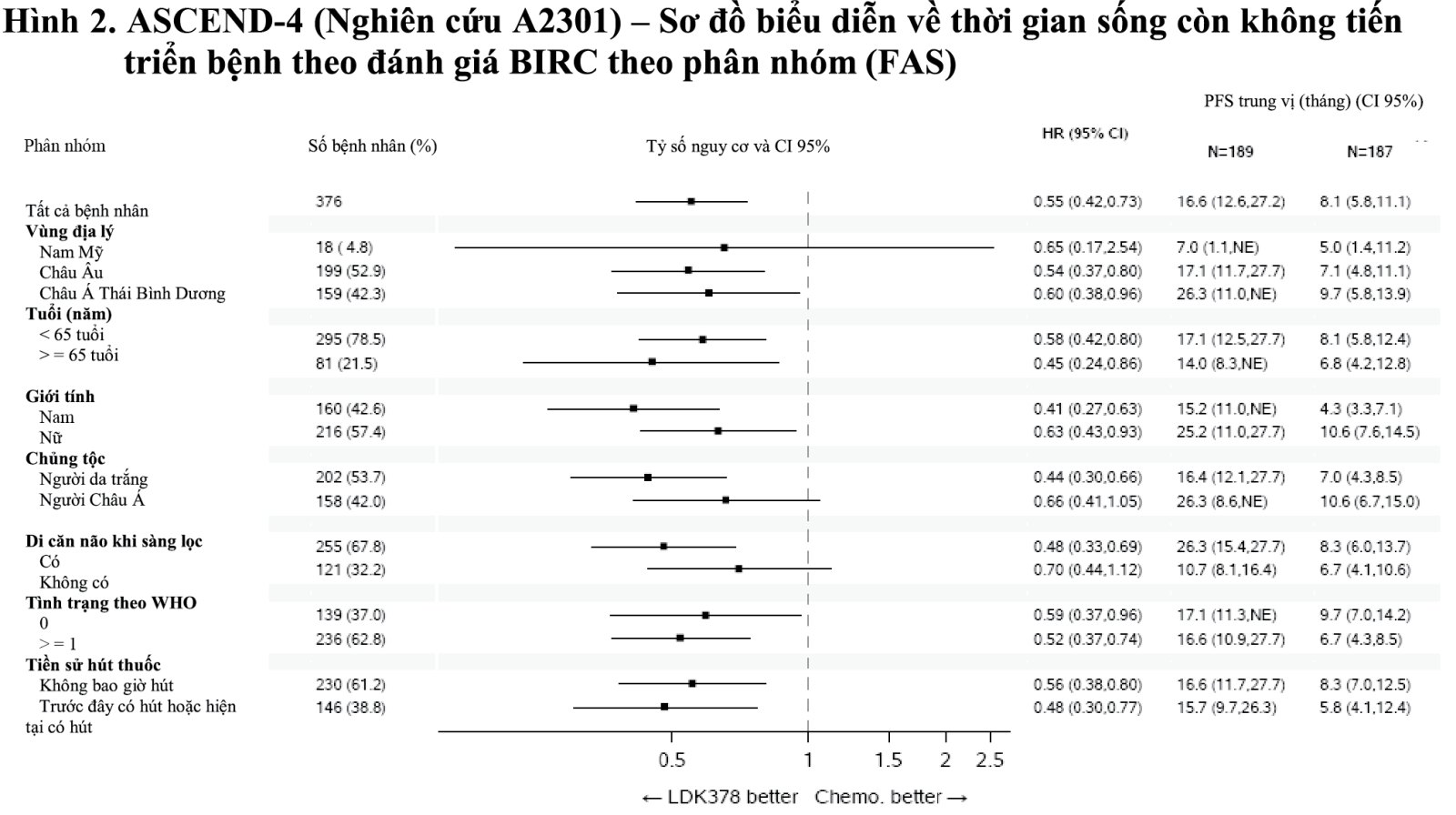
Như được xác định trước trong đề cương nghiên cứu, thời gian sống còn toàn bộ (OS) được kiểm định chính thức dưới dạng thời gian sống còn không tiến triển bệnh (PFS) là tiêu chí hiệu quả chính theo đánh giá của BIRC có ý nghĩa thống kê và thiên về nhóm ceritinib. Dữ liệu về thời gian sống còn toàn bộ (OS) chưa hoàn thiện với 107 biến cố tử vong tương đương khoảng 42,3% biến cố cần thiết cho phân tích cuối cùng về thời gian sống còn toàn bộ. Có ít biến cố tử vong ở nhóm ceritinib (48 biến cố, 25,4%) hơn so với nhóm hóa trị liệu (59 biến cố, 31,6%), cho thấy xu hướng ưu thế ceritinib (tỷ số nguy cơ (HR): 0,73 với khoảng tin cậy (CI) 95%: 0,50; 1,08, kiểm định phân tầng log-rank một phía p=0,056). Thời gian sống còn toàn bộ trung vị không ước tính được (NE) ở nhóm ceritinib và là 26,2 tháng (khoảng tin cậy (CI) 95%: 22,8, NE) ở nhóm hóa trị liệu. Tỷ lệ sống còn toàn bộ ước tính (khoảng tin cậy (CI) 95%) lúc 24 tháng là 70,6% (62,2; 77,5) đối với nhóm ceritinib và là 58,2% (47,6; 67,5) đối với nhóm hóa trị liệu. Tám mươi mốt bệnh nhân (43,3%) ở nhóm hóa trị liệu dùng ceritinib sau đó như là liệu pháp chống ung thư đầu tiên sau khi ngừng điều trị nghiên cứu.
Dữ liệu về hiệu quả từ nghiên cứu A2301 được tóm tắt trong Bảng 1 và các đường cong Kaplan-Meier về thời gian sống còn không tiến triển bệnh (PFS) và thời gian sống còn toàn bộ (OS) và sơ đồ biểu diễn về thời gian sống còn không tiến triển bệnh theo phân nhóm được thể hiện trong Hình 1, Hình 2 và Hình 3.
- xem Bảng 1, Hình 1, Hình 2
Ngoại trừ “tình trạng theo Tổ chức Y tế Thế giới (WHO)” và “Di căn não khi sàng lọc”, mô hình hồi quy Cox được phân tầng bởi sự hiện diện hoặc không có di căn não, hóa trị liệu bổ trợ trước đó và tình trạng theo WHO.
Đối với “tình trạng theo WHO”, mô hình hồi quy Cox được phân tầng bởi sự có hoặc không có di căn não, hóa trị liệu bổ trợ trước đó.
Đối với “di căn não khi sàng lọc”, mô hình hồi quy Cox được phân tầng theo tình trạng của WHO và sự hiện diện hoặc không có hóa trị liệu bổ trợ trước đó.
- xem Hình 3.
Các bảng câu hỏi kết quả do bệnh nhân báo cáo đã được hoàn thành bởi 80% bệnh nhân hoặc hơn ở nhóm ceritinib và nhóm hóa trị liệu đối với tất cả các bảng câu hỏi ở hầu hết các thời điểm trong quá trình nghiên cứu.
Ceritinib kéo dài đáng kể thời gian đến khi giảm các triệu chứng đặc hiệu của ung thư phổi như thể hiện bởi tiêu chí tổng hợp gồm ho, đau và khó thở bằng công cụ điểm số triệu chứng ung thư phổi (LCSS) (Tỷ số nguy cơ (HR) = 0,61, khoảng tin cậy (CI) 95%: 0,41; 0,90) và QLQ-LC13 (Bảng câu hỏi chất lượng cuộc sống trong ung thư phổi) (Tỷ số nguy cơ (HR) = 0,48, khoảng tin cậy (CI) 95%: 0,34; 0,69) so với hóa trị liệu. Thời gian trung vị đến khi giảm rõ rệt đối với tiêu chí tổng hợp về LC13 (đau, ho, khó thở) là 23,6 tháng (khoảng tin cậy (CI) 95%: 20,7; không đạt được) ở nhóm ceritinib so với 12,6 tháng (khoảng tin cậy (CI) 95%: 8,9; 14,9) ở nhóm hóa trị liệu.
Những bệnh nhân dùng ceritinib đã cho thấy những sự cải thiện có ý nghĩa so với hóa trị liệu theo thang điểm chất lượng cuộc sống nói chung (LCSS, p<0,001), tình trạng sức khỏe tổng thể/chất lượng cuộc sống (QoL) (QLQ-C30, p<0,001) cũng như về chỉ số EQ-5D-5L (p<0,001) và EQ-5D-5L VAS (p<0,05 từ mỗi chu kỳ điều trị từ 13 đến 49). Nhìn chung, các kết quả này cho thấy những sự cải thiện về các triệu chứng đặc hiệu ung thư phổi cũng như lợi ích về tình trạng sức khỏe chung đối với bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC), ALK dương tính được điều trị bằng ceritinib so với hóa trị liệu.
Trong nghiên cứu A2301, 44 trong số 121 bệnh nhân bị di căn não hoạt động và có thể đánh giá được lúc ban đầu và ít nhất một đánh giá bằng chụp X-quang não sau lúc ban đầu (22 bệnh nhân ở nhóm ceritinib và 22 bệnh nhân ở nhóm hóa trị liệu). Di căn não hoạt động được định nghĩa là di căn não không được điều trị, di căn não mới hoặc di căn não với sự tiến triển được ghi nhận sau khi kết thúc chương trình xạ trị não. 44 bệnh nhân này đã được đánh giá về đáp ứng nội sọ bởi các bác sĩ chuyên khoa X-quang thần kinh của BIRC. Tỷ lệ đáp ứng nội sọ toàn bộ (OIRR) cao hơn với ceritinib (72,7%, khoảng tin cậy (CI): 49,8; 89,3) so với nhóm hóa trị liệu (27,3%, khoảng tin cậy (CI) 95%: 10,7; 50,2). Trong số những bệnh nhân bị di căn não có thể đánh giá được lúc ban đầu và ít nhất một đánh giá sau lúc ban đầu, 59,1% (13/22) ở nhóm ceritinib và 81,8% (18/22) ở nhóm hóa trị liệu đã không nhận được xạ trị não trước đó.
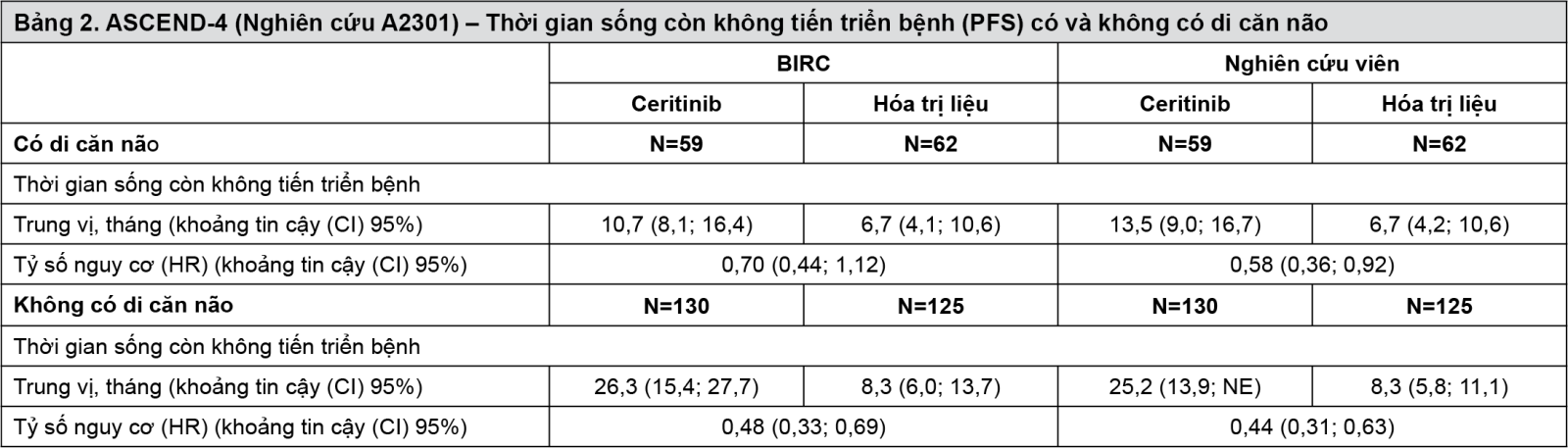
Thời gian sống còn không tiến triển bệnh (PFS) trung vị theo đánh giá của BIRC và nghiên cứu viên sử dụng RECIST 1.1 dài hơn ở nhóm ceritinib so với nhóm hóa trị liệu ở cả hai phân nhóm bệnh nhân có di căn não và không có di căn não (dựa trên mức độ mệt mỏi liên quan đến ung thư (CRF), xem Bảng 2).
- xem Bảng 2.
Ung thư phổi không tế bào nhỏ (NSCLC) tiến xa tại chỗ hoặc di căn, dương tính với ALK, được điều trị trước đây - Nghiên cứu A2303 ngẫu nhiên pha 3 (ASCEND-5)
Hiệu quả và độ an toàn của Spexib trong điều trị bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC), dương tính với ALK, tiến xa tại chỗ hoặc di căn, có và không có di căn não, đã được điều trị trước đây bằng crizotinib, đã được chứng minh trong nghiên cứu A2303 pha 3, đa trung tâm toàn cầu, ngẫu nhiên, nhãn mở.
Tiêu chí hiệu quả chính là thời gian sống còn không tiến triển bệnh (PFS), như được xác định bởi Ủy ban xem xét độc lập, mù với phân bổ điều trị (BIRC), theo tiêu chuẩn đánh giá đáp ứng về khối u đặc (RECIST) 1.1. Tiêu chí phụ quan trọng là thời gian sống còn toàn bộ (OS). Các tiêu chí phụ khác bao gồm tỷ lệ đáp ứng toàn bộ (ORR), thời gian đáp ứng (DOR), tỷ lệ kiểm soát bệnh (DCR) và thời gian đến khi đáp ứng (TTR) được xác định bởi BIRC và nghiên cứu viên và kết quả do bệnh nhân báo cáo (PRO), bao gồm các triệu chứng liên quan đến bệnh, chức năng và chất lượng cuộc sống liên quan đến sức khỏe.
Tỷ lệ đáp ứng nội sọ toàn bộ (OIRR), tỷ lệ kiểm soát bệnh nội sọ (IDCR) và thời gian đến khi đáp ứng nội sọ (DOIR) được xác định bởi bác sĩ chuyên khoa X-quang thần kinh của BIRC theo RECIST 1.1 sửa đổi (tức là lên đến 5 tổn thương trong não) đã được sử dụng để đánh giá hoạt tính chống khối u trong não.
Các bệnh nhân được phép tiếp tục điều trị nghiên cứu được phân bổ sau tiến triển ban đầu trong trường hợp tiếp tục có lợi ích lâm sàng theo ý kiến của nghiên cứu viên. Các bệnh nhân được phân ngẫu nhiên vào nhóm hóa trị liệu có thể bắt chéo để dùng ceritinib dựa trên sự tiến triển bệnh được xác định bởi BIRC theo RECIST.
Tổng cộng có 231 bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC) tiến xa, dương tính với ALK đã được điều trị trước đó bằng crizotinib và hóa trị liệu (một hoặc hai phác đồ bao gồm một bộ đôi dựa trên platin) đã được đưa vào phân tích. 115 bệnh nhân được chọn ngẫu nhiên để dùng ceritinib và 116 bệnh nhân được chọn ngẫu nhiên để dùng hóa trị liệu (pemetrexed hoặc docetaxel). 73 bệnh nhân dùng docetaxel và 40 bệnh nhân dùng pemetrexed. Ở nhóm ceritinib, 115 bệnh nhân đã được điều trị.
Đặc điểm bệnh ban đầu rất đồng đều giữa hai nhóm điều trị. Độ tuổi trung vị là 54 tuổi (từ 28-84 tuổi); 77,1% bệnh nhân trẻ hơn 65 tuổi. Tổng cộng có 55,8% bệnh nhân là nữ. 64,5% nhóm đối tượng nghiên cứu là người da trắng, 29,4% là người châu Á, 0,4% là người da đen và 2,6% là các chủng tộc khác. Đa số bệnh nhân bị ung thư tuyến (97,0%) và chưa bao giờ hút thuốc lá hoặc đã từng hút thuốc lá trước đây (96,1%). Tình trạng hoạt động cơ thể theo thang điểm của Nhóm hợp tác ung thư miền đông (ECOG) theo thứ tự là 0/1/2 ở 46,3%/47,6%/6,1% bệnh nhân, và 58,0% có di căn não lúc ban đầu. Tất cả các bệnh nhân đã được điều trị bằng crizotinib trước đó. 198 bệnh nhân (81,8%) dùng crizotinib như điều trị cuối cùng (81,7% ở nhóm ceritinib, 81,9% ở nhóm hóa trị liệu). Tất cả trừ một bệnh nhân đã dùng hóa trị liệu trước đó (bao gồm một bộ đôi dựa trên platin) đối với bệnh tiến triển; 11,3% bệnh nhân ở nhóm ceritinib và 12,1% bệnh nhân ở nhóm hóa trị liệu đã được điều trị bằng hai phác đồ hóa trị liệu đối với bệnh tiến triển.
Thời gian theo dõi trung vị là 16,5 tháng (từ ngày chọn ngẫu nhiên đến ngày khóa dữ liệu).
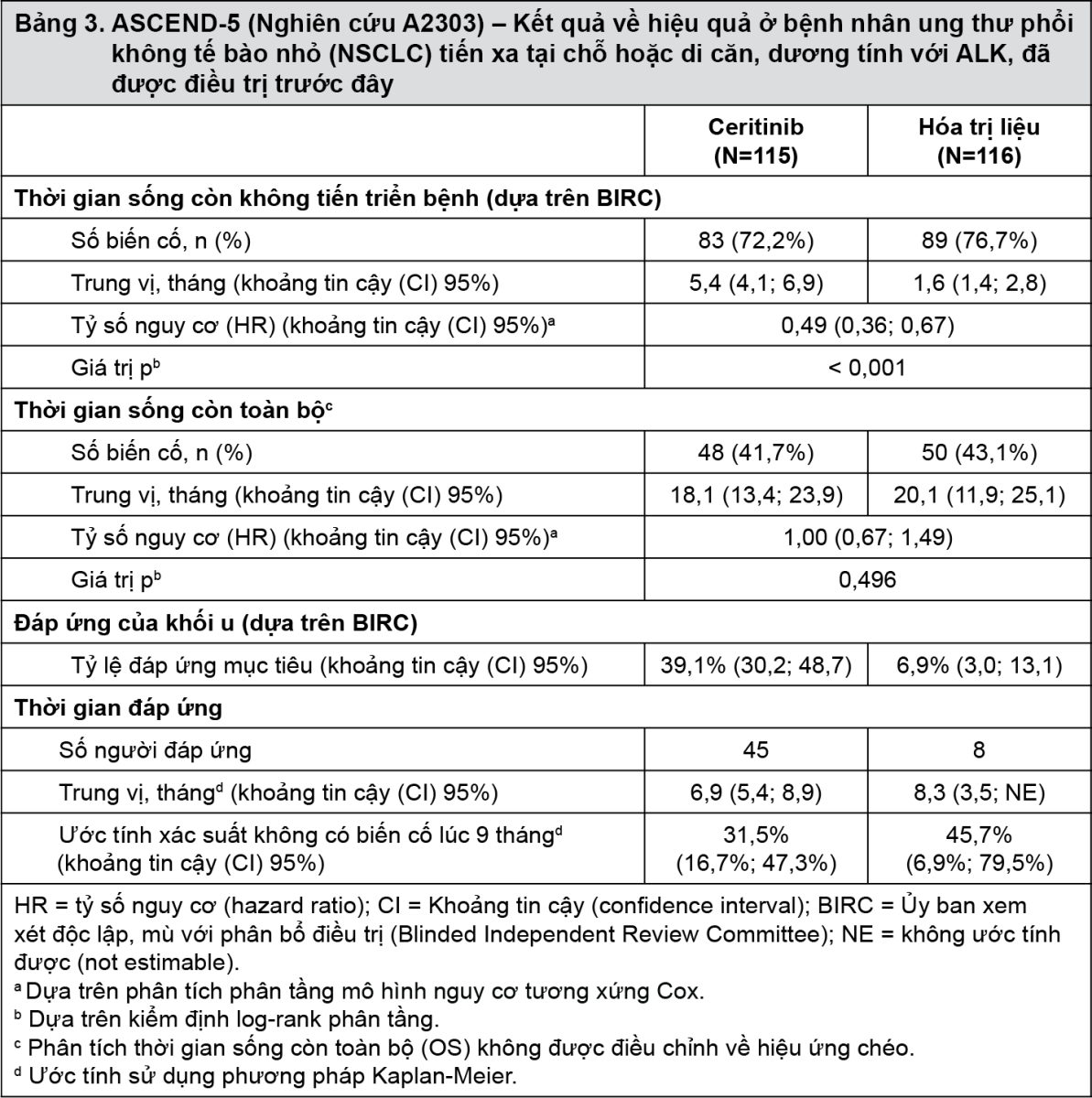
Nghiên cứu này đã đáp ứng mục tiêu chính cho thấy sự cải thiện có ý nghĩa thống kê và có ý nghĩa lâm sàng về thời gian sống còn không tiến triển bệnh (PFS) theo đánh giá của BIRC với sự giảm nguy cơ ước tính là 51% ở nhóm ceritinib so với nhóm hóa trị liệu (Tỷ số nguy cơ (HR): 0,49 với khoảng tin cậy (CI) 95%: 0,36; 0,67). Thời gian sống còn không tiến triển bệnh (PFS) trung vị là 5,4 tháng (khoảng tin cậy (CI) 95%: 4,1; 6,9) đối với nhóm ceritinib và là 1,6 tháng (khoảng tin cậy (CI) 95%: 1,4; 2,8) đối với nhóm hóa trị liệu (xem Bảng 3 và Hình 4).
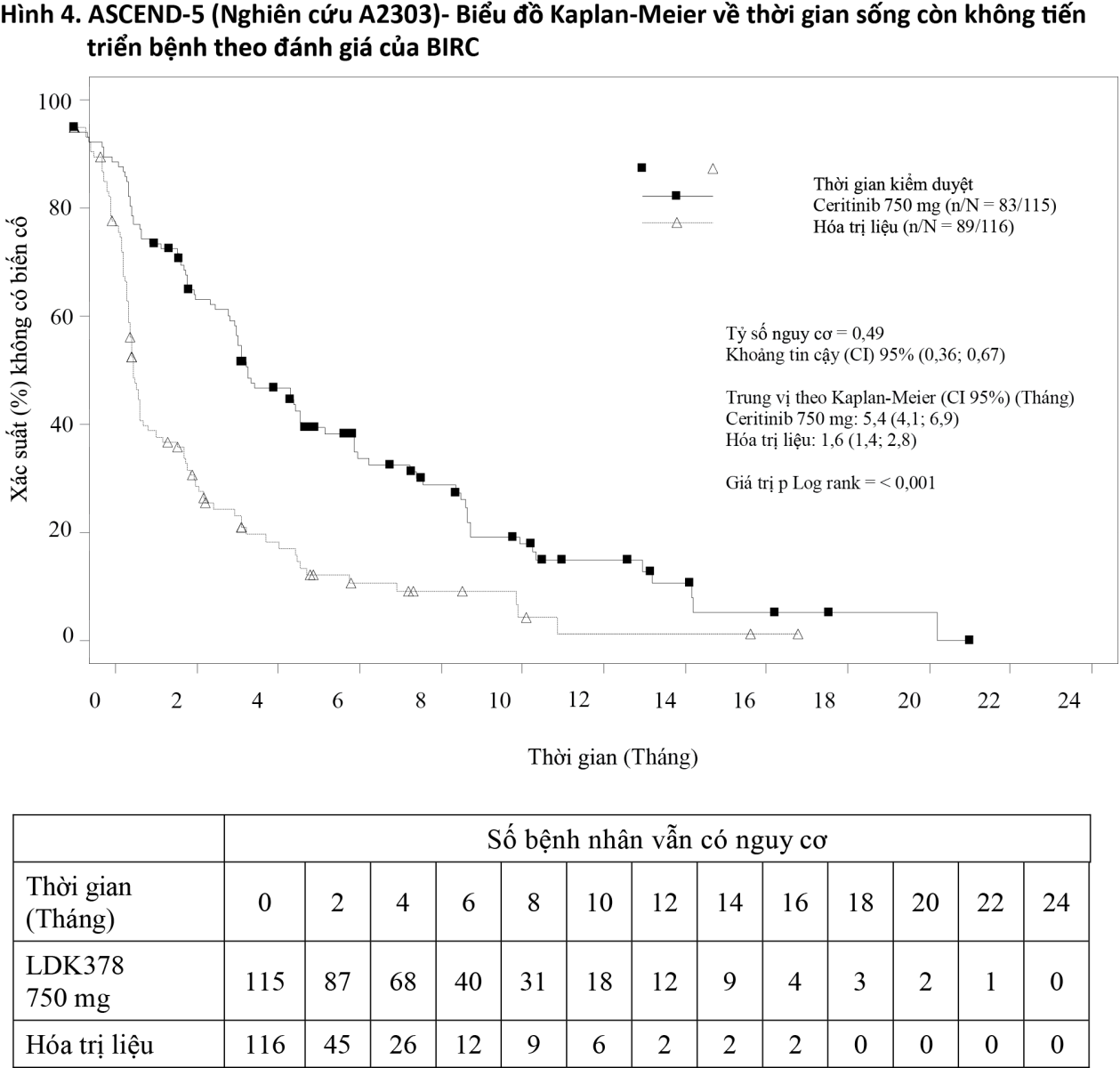
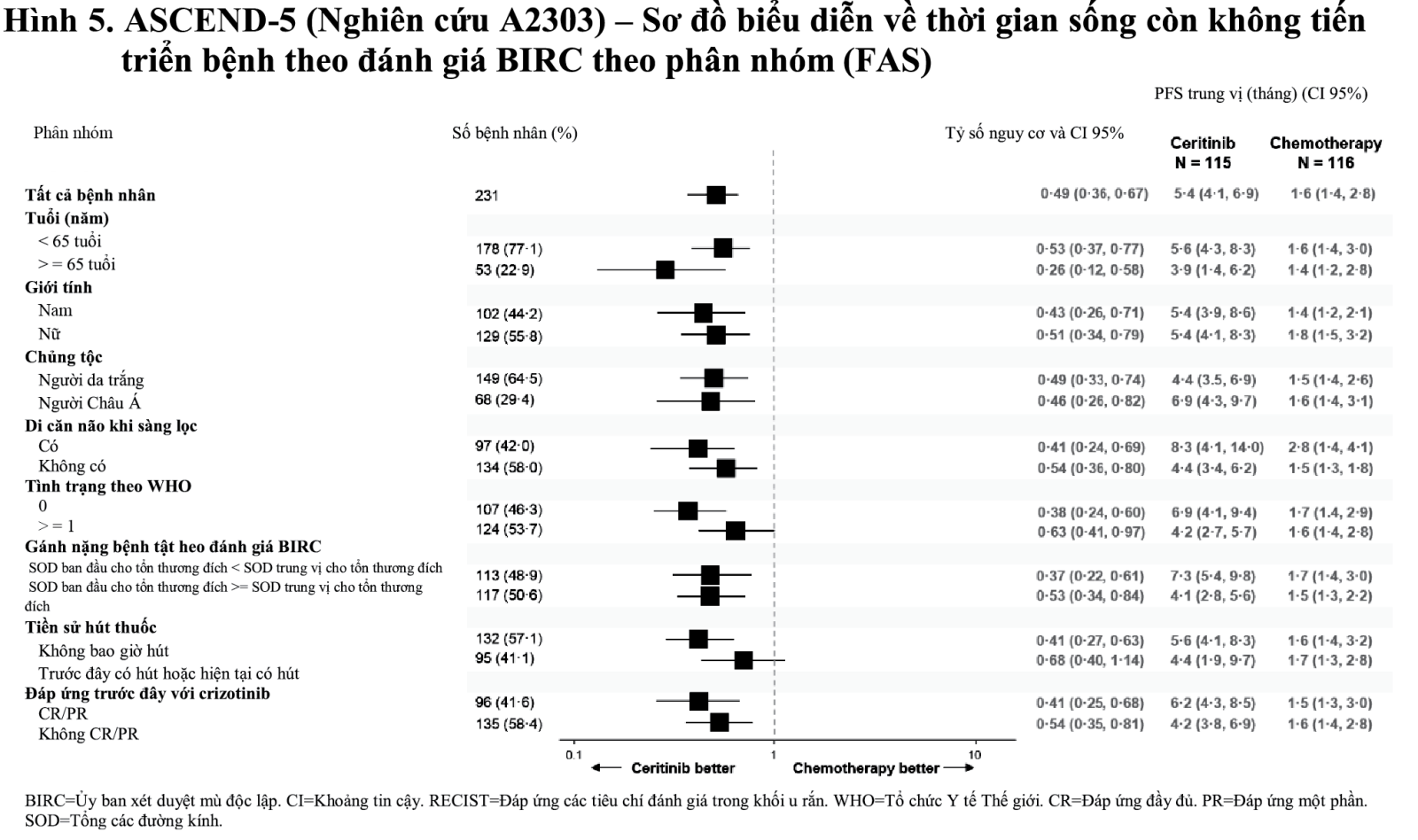
Lợi ích của ceritinib về thời gian sống còn không tiến triển bệnh (PFS) vượt trội so với hóa trị liệu là rõ ràng và đồng nhất theo đánh giá của nghiên cứu viên và ở các phân nhóm khác nhau bao gồm tuổi, giới tính, chủng tộc, nhóm hút thuốc lá, tình trạng hoạt động cơ thể theo thang điểm của ECOG, sự hiện diện của di căn não hoặc đáp ứng trước đó với crizotinib (xem Hình 5).
Lợi ích đã được hỗ trợ thêm bằng phân tích tỷ lệ đáp ứng toàn bộ (ORR) và tỷ lệ kiểm soát bệnh (DCR). Ceritinib cũng cải thiện có ý nghĩa tỷ lệ đáp ứng toàn bộ (ORR) theo đánh giá của BIRC so với hóa trị liệu với đáp ứng lâu bền (xem Bảng 3).
Như được xác định trước trong đề cương, thời gian sống còn toàn bộ (OS) được kiểm định chính thức dưới dạng thời gian sống còn không tiến triển bệnh (PFS) là tiêu chí hiệu quả chính theo đánh giá của BIRC có ý nghĩa thống kê và ưu thế cho nhóm ceritinib. Dữ liệu về thời gian sống còn toàn bộ (OS) chưa hoàn thiện với 48 biến cố (41,7%) ở nhóm ceritinib và 50 biến cố (43,1%) ở nhóm hóa trị liệu, tương ứng với khoảng 50% biến cố cần thiết cho việc phân tích cuối cùng về thời gian sống còn toàn bộ. Ngoài ra, 81 bệnh nhân (69,8%) ở nhóm hóa trị liệu dùng ceritinib sau đó dưới dạng liệu pháp chống ung thư đầu tiên sau khi ngừng điều trị nghiên cứu.
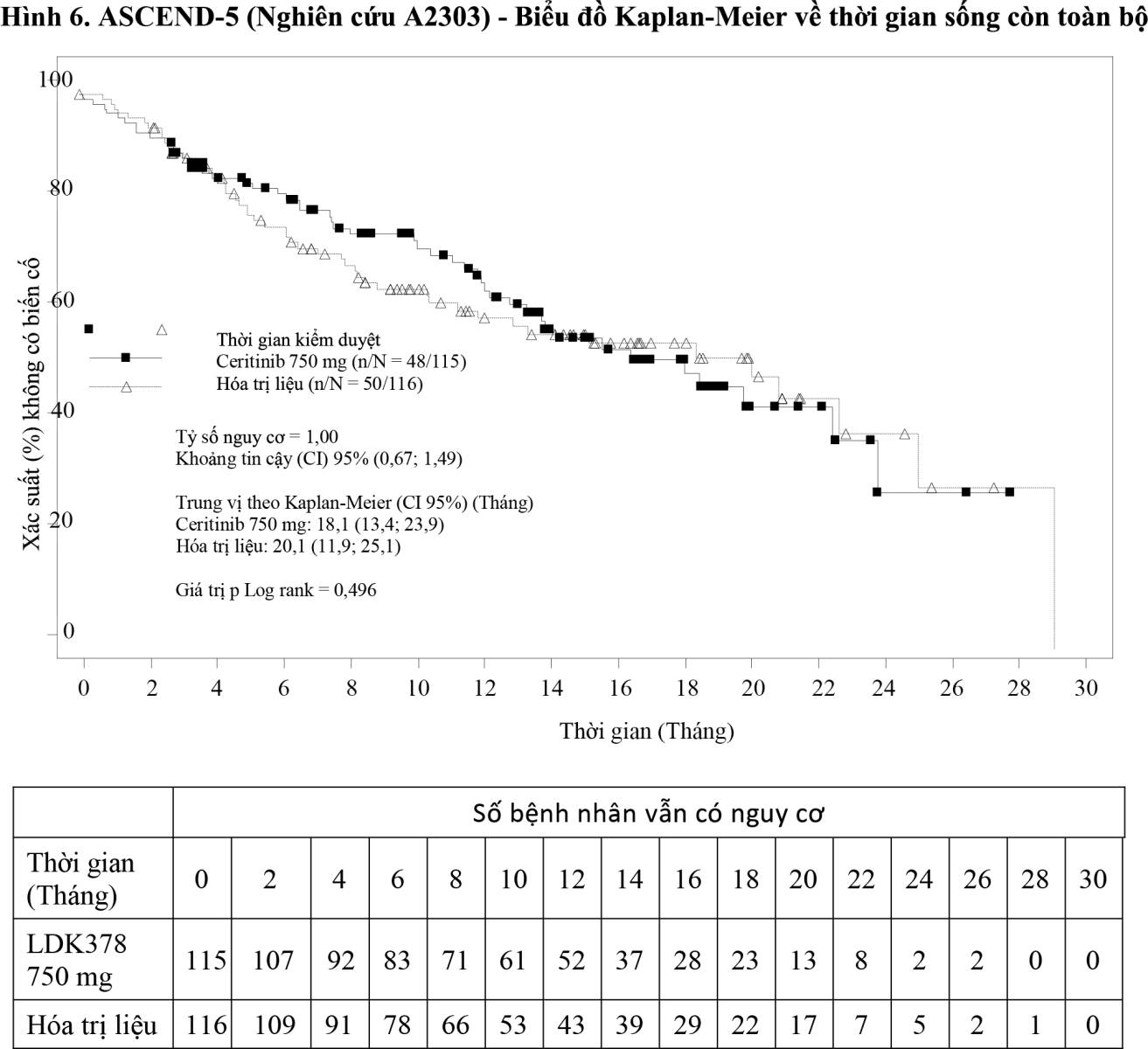
Dữ liệu về hiệu quả từ nghiên cứu A2303 được tóm tắt trong Bảng 3 và các đường cong Kaplan-Meier về thời gian sống còn không tiến triển bệnh (PFS) và thời gian sống còn toàn bộ (OS) và sơ đồ biểu diễn về thời gian sống còn không tiến triển bệnh theo phân nhóm được thể hiện trong Hình 4, Hình 5 và Hình 6.
- xem Bảng 3, Hình 4, Hình 5.
Ngoại trừ “Tình trạng theo WHO” và “Di căn não khi sàng lọc”, tỷ số nguy cơ dựa trên mô hình hồi quy Cox được phân tầng bởi sự hiện diện hoặc không có di căn não và tình trạng theo WHO theo IRT ngẫu nhiên.
Di căn não lúc sàng lọc theo dữ liệu CRF lúc ban đầu.
Đối với “di căn não khi sàng lọc”, mô hình hồi quy Cox được phân tầng theo tình trạng của WHO theo ngẫu nhiên (IRT).
Đối với “tình trạng theo WHO”, mô hình hồi quy Cox được phân tầng bởi sự có mặt hoặc không di căn não theo ngẫu nhiên (IRT).
Phân nhóm của những người hút thuốc lá trước đây hoặc người đang hút thuốc lá bao gồm 5 bệnh nhân đang hút thuốc.
- xem Hình 6.
Các bảng câu hỏi kết quả do bệnh nhân báo cáo đã được hoàn thành bởi 75% bệnh nhân hoặc hơn ở nhóm ceritinib và nhóm hóa trị liệu đối với tất cả các bảng câu hỏi ở hầu hết các thời điểm trong quá trình nghiên cứu.
Sự cải thiện có ý nghĩa đã được báo cáo đối với đa số các triệu chứng đặc hiệu của ung thư phổi đối với Spexib so với hóa trị liệu (điểm số triệu chứng ung thư phổi (LCSS) và QLQ-LC13 (Bảng câu hỏi chất lượng cuộc sống trong ung thư phổi)). Thời gian đến khi giảm rõ rệt đối với ho, đau và khó thở đã được kéo dài có ý nghĩa đối với từng thang điểm riêng lẻ (giá trị p<0,05) hoặc khi kết hợp thành một điểm số tổng hợp (giá trị p<0,001) trong các công cụ LCSS và LC13. Thời gian trung vị đến khi giảm rõ rệt đối với các tiêu chí tổng hợp về LCSS (đau, ho, khó thở) là 18 tháng (khoảng tin cậy (CI) 95%: 13,4, NE) ở nhóm ceritinib so với 4,4 tháng (khoảng tin cậy (CI) 95%: 1,6; 8,6) ở nhóm hóa trị liệu. Thời gian trung vị đến khi giảm rõ rệt đối với cùng tiêu chí về LC13 là 11,1 tháng (khoảng tin cậy (CI) 95%: 7,1; 14,2) ở nhóm ceritinib so với 2,1 tháng (khoảng tin cậy (CI) 95%: 1,0; 5,6) ở nhóm hóa trị liệu.
Bảng câu hỏi EQ-5D đã cho thấy sự cải thiện tình trạng sức khỏe tổng thể có ý nghĩa đối với Spexib so với hóa trị liệu.
Trong nghiên cứu A2303, 133 bệnh nhân có di căn não lúc ban đầu (66 bệnh nhân ở nhóm ceritinib và 67 bệnh nhân ở nhóm hóa trị liệu) đã được đánh giá về đáp ứng nội sọ bởi bác sĩ chuyên khoa X-quang thần kinh. Tỷ lệ đáp ứng nội sọ toàn bộ (OIRR) ở những bệnh nhân bị bệnh ở não có thể đánh giá được lúc ban đầu và ít nhất một đánh giá sau lúc ban đầu cao hơn với ceritinib (35,3%, khoảng tin cậy (CI) 95%: 14,2; 61,7) so với hóa trị liệu (5,0%, khoảng tin cậy (CI) 95%: 0,1; 24,9).
Thời gian sống còn không tiến triển bệnh (PFS) trung vị theo đánh giá của BIRC và nghiên cứu viên sử dụng RECIST 1.1 dài hơn ở nhóm ceritinib so với nhóm hóa trị liệu ở cả hai phân nhóm bệnh nhân có di căn não và không có di căn não (dựa trên mức độ mệt mỏi liên quan đến ung thư (CRF), xem Bảng 4).
- xem Bảng 4.
Các nghiên cứu đơn nhóm X2101, A2203 và A2201
Sử dụng Spexib trong điều trị cho bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK được đánh giá trong 3 thử nghiệm lâm sàng toàn cầu, đa trung tâm, nhãn mở, đơn nhóm (Nghiên cứu X2101, Nghiên cứu A2203 và Nghiên cứu A2201).
Tiêu chí đánh giá chính về hiệu quả của thuốc trong các nghiên cứu này là tỷ lệ đáp ứng toàn bộ (ORR) trên bệnh nhân điều trị bằng Spexib liều 750 mg, được định nghĩa là tỷ lệ bệnh nhân có đáp ứng tốt nhất trong các bệnh nhân đáp ứng hoàn toàn (CR) hoặc đáp ứng một phần (PR) được khẳng định bằng các đánh giá lặp lại thực hiện không dưới 4 tuần sau lần đầu tiên đạt được tiêu chí đáp ứng. Các đánh giá bổ sung bao gồm thời gian đáp ứng (DOR) và thời gian sống thêm không bệnh tiến triển (PFS) theo đánh giá của nghiên cứu viên và thời gian sống thêm toàn bộ (OS). Các đánh giá về khối u được thực hiện bởi các nghiên cứu viên theo tiêu chí đánh giá đáp ứng trên các khối u đặc (RECIST) 1.0 trong Nghiên cứu X2101 và RECIST 1.1 trong Nghiên cứu A2203 và A2201.
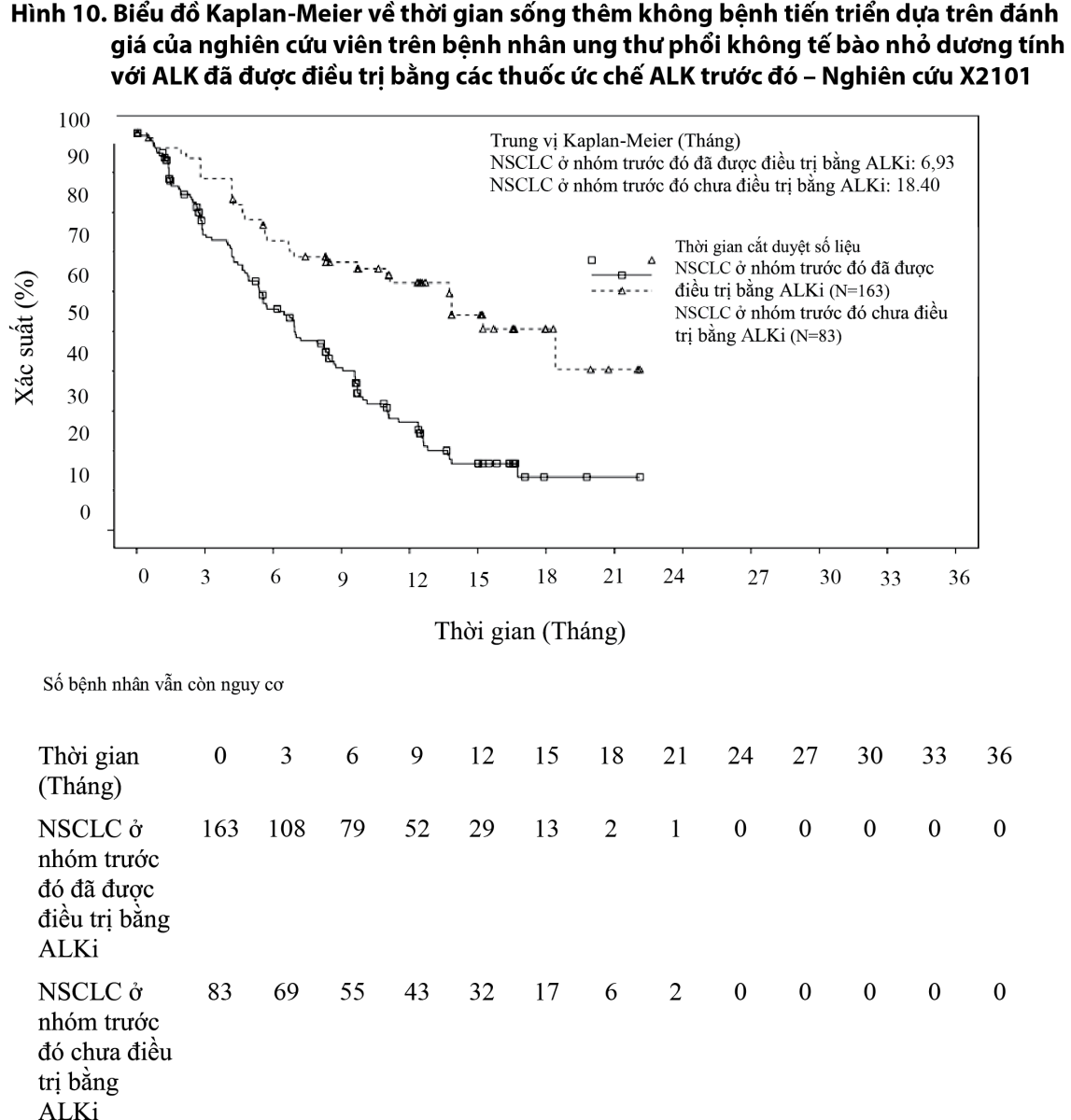
Nghiên cứu X2101 là một nghiên cứu toàn cầu, đa trung tâm, nhãn mở, pha 1 bao gồm giai đoạn dò liều tăng dần và giai đoạn mở rộng, ở mức liều được khuyến cáo là 750 mg. Tất cả bệnh nhân tham gia vào nghiên cứu có khối u tiến xa tại chỗ hoặc di căn tiến triển mặc dù đã được điều trị theo phác đồ chuẩn và tất cả bệnh nhân trước đây đã được xét nghiệm về sự chuyển vị ALK. Bệnh nhân bị di căn não đã được kiểm soát hoặc không biểu hiện triệu chứng được lựa chọn tham gia nghiên cứu. Điều trị bằng thuốc ức chế ALK trước đó được cho phép. 290 trong số 304 bệnh nhân tham gia vào nghiên cứu là các bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK. Tổng số 246 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK tham gia vào nghiên cứu được điều trị bằng Spexib với liều 750 mg: 163 bệnh nhân trước đó đã được điều trị bằng thuốc ức chế ALK và 83 bệnh nhân chưa từng dùng thuốc ức chế ALK.
Trong 246 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK được điều trị với mức liều 750 mg trong nghiên cứu, tuổi trung vị là 53 tuổi (dao động từ 22 đến 80 tuổi); 84,1% bệnh nhân dưới 65 tuổi. 53,7% bệnh nhân là nữ. Bệnh nhân da trắng chiếm 63,4% quần thể nghiên cứu, bệnh nhân là người châu Á chiếm 33,3%, người da đen chiếm 1,6% và các chủng tộc khác chiếm 1,6%. Phần lớn các bệnh nhân bị carcinôm tuyến (92,7%), các bệnh nhân này đã hoặc chưa từng hút thuốc (97,6%). Hơn hai phần ba (67,5%) bệnh nhân tham gia vào nghiên cứu trước đó đã được điều trị từ 2 phác đồ trở lên, 26,0% bệnh nhân đã được điều trị bằng 1 phác đồ trước đó và 6,5% bệnh nhân chưa được điều trị trước đó.
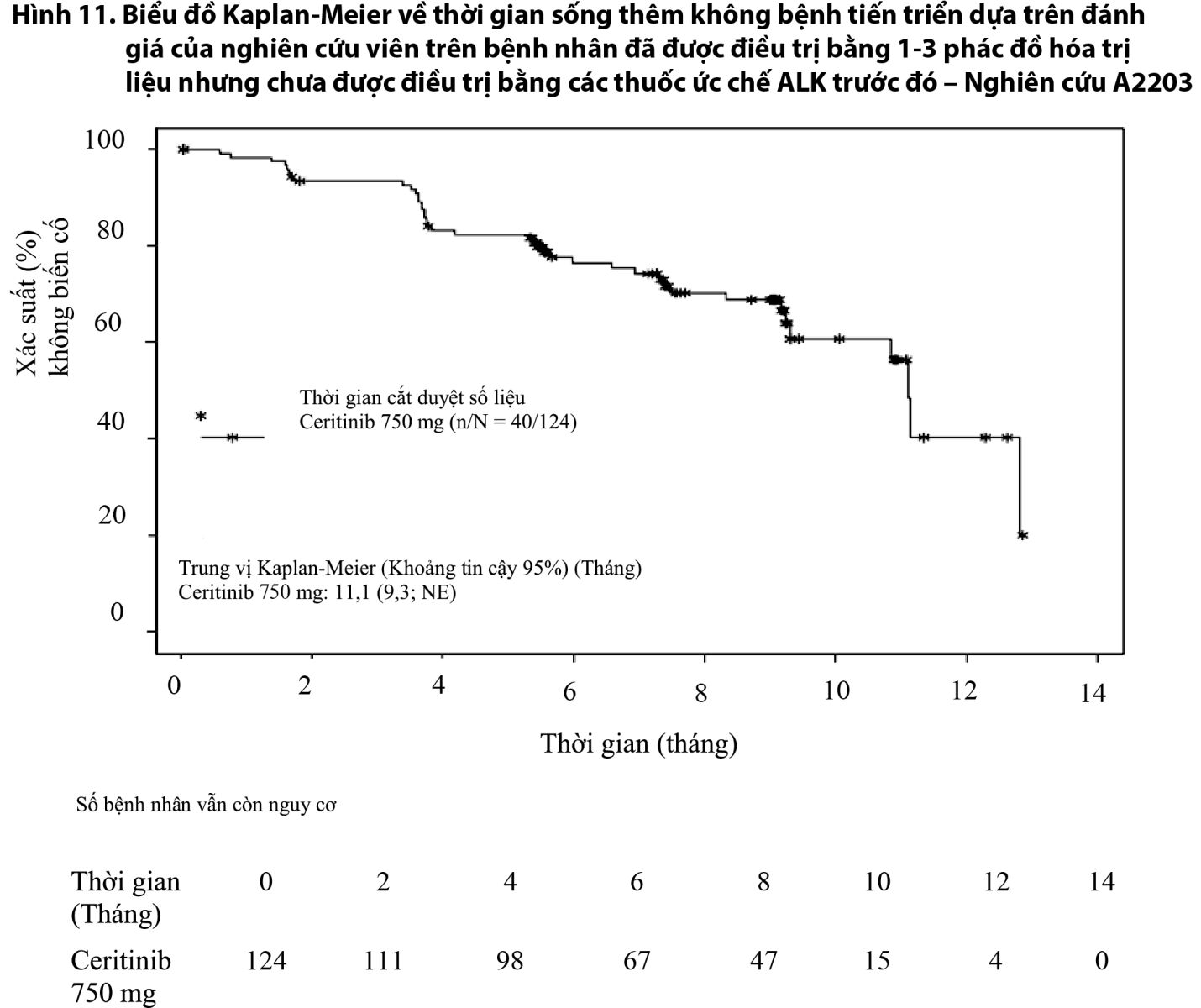
Nghiên cứu A2203 và Nghiên cứu A2201 đều là nghiên cứu toàn cầu, đa trung tâm, nhãn mở, đơn nhóm, pha 2, được thiết kế để đánh giá hiệu quả và độ an toàn của ceritinib 750 mg trên bệnh nhân ung thư phổi không tế bào nhỏ tiến triển tại chỗ hoặc có di căn dương tính với ALK. Nghiên cứu A2203 bao gồm 124 bệnh nhân chưa dùng crizotinib trước đó, các bệnh nhân này hoặc chưa dùng hóa trị liệu hoặc đã được điều trị từ 1-3 phác đồ hóa trị liệu trước đó. Nghiên cứu A2201 bao gồm 140 bệnh nhân trước đó đã được điều trị từ 1-3 phác đồ hóa trị liệu, sau đó được điều trị bằng crizotinib và có bệnh tiến triển trong quá trình điều trị bằng crizotinib.
Trong Nghiên cứu A2203, 124 bệnh nhân được điều trị ở mức liều 750 mg. Tuổi trung vị của các bệnh nhân này là 56 tuổi (dao động từ 27 đến 82 tuổi); 75,8% bệnh nhân dưới 65 tuổi. Có 59,7% bệnh nhân là nữ giới. Người châu Á chiếm 59,7% quần thể nghiên cứu, người da trắng chiếm 38,7%, người da đen chiếm 0,8% và các chủng tộc khác 0,8%. Phần lớn bệnh nhân bị carcinôm tuyến (96,8%). Tất cả bệnh nhân, ngoại trừ 2 người trước đó chưa từng dùng thuốc chống ung thư, đã được điều trị bằng các phác đồ hóa trị liệu trước đó: 54 (43,5%) bệnh nhân đã được điều trị bằng 1 phác đồ và 68 bệnh nhân (54,8%) đã được điều trị từ 2 phác đồ trở lên trước khi tham gia vào nghiên cứu. Tất cả các bệnh nhân đều chưa từng dùng thuốc ức chế ALK.
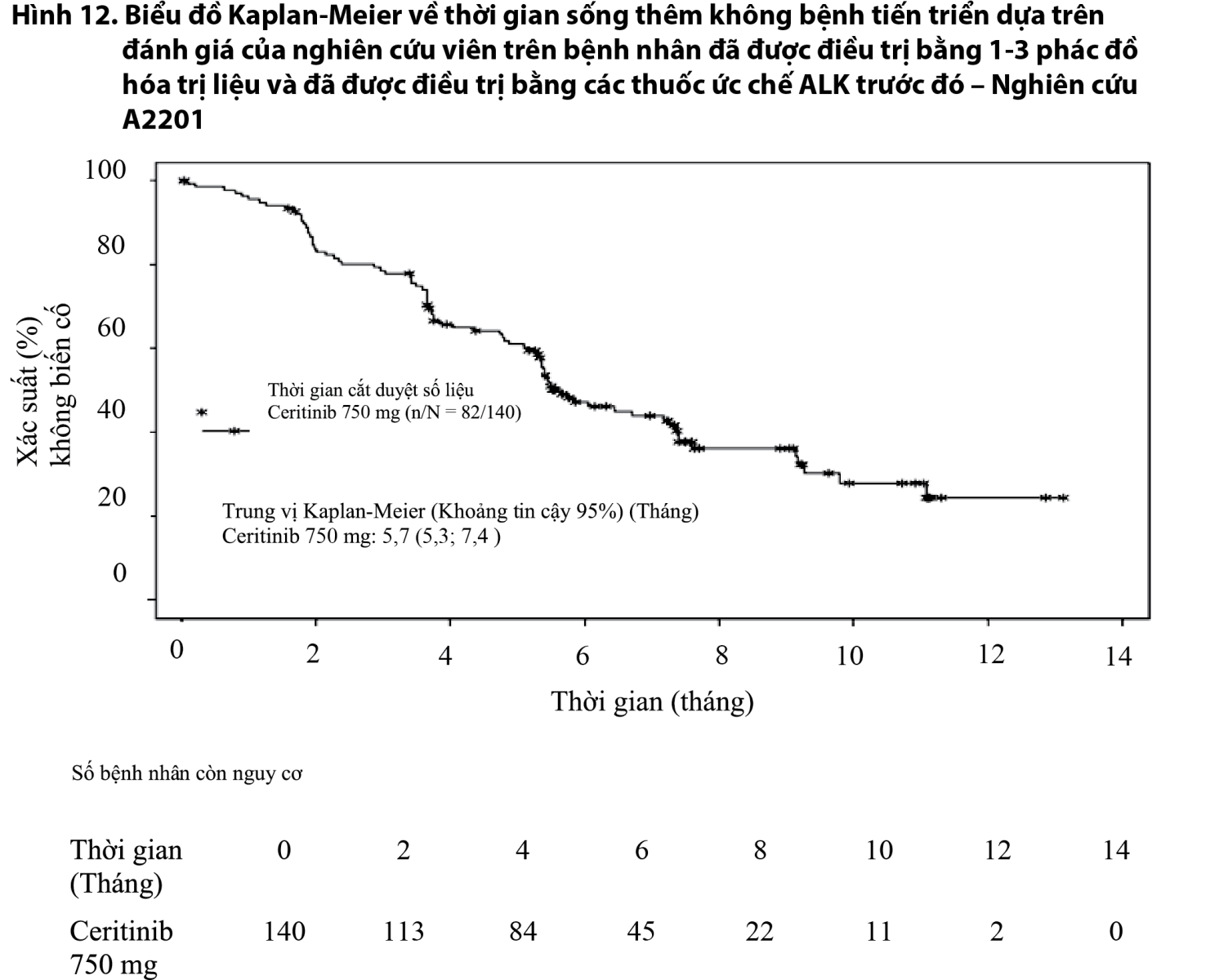
Trong nghiên cứu A2201, 140 bệnh nhân được điều trị với mức liều 750 mg. Tuổi trung vị của bệnh nhân là 51 tuổi (dao động từ 29 đến 80 tuổi); 87,1% bệnh nhân dưới 65 tuổi. Có 50% số bệnh nhân là nữ giới. Người da trắng chiếm 60% quần thể nghiên cứu, người châu Á chiếm 37,9% và các chủng tộc khác 2,1%. Đại đa số các bệnh nhân bị carcinôm tuyến (92,1%). Tất cả các bệnh nhân đã được điều trị từ 2 phác đồ hóa trị liệu trở lên trước khi tham gia vào nghiên cứu. Tất cả các bệnh nhân trước đó đã được điều trị bằng thuốc ức chế ALK.
Các kết quả chính đánh giá hiệu quả của thuốc trong các Nghiên cứu X2101, A2203 và A2201
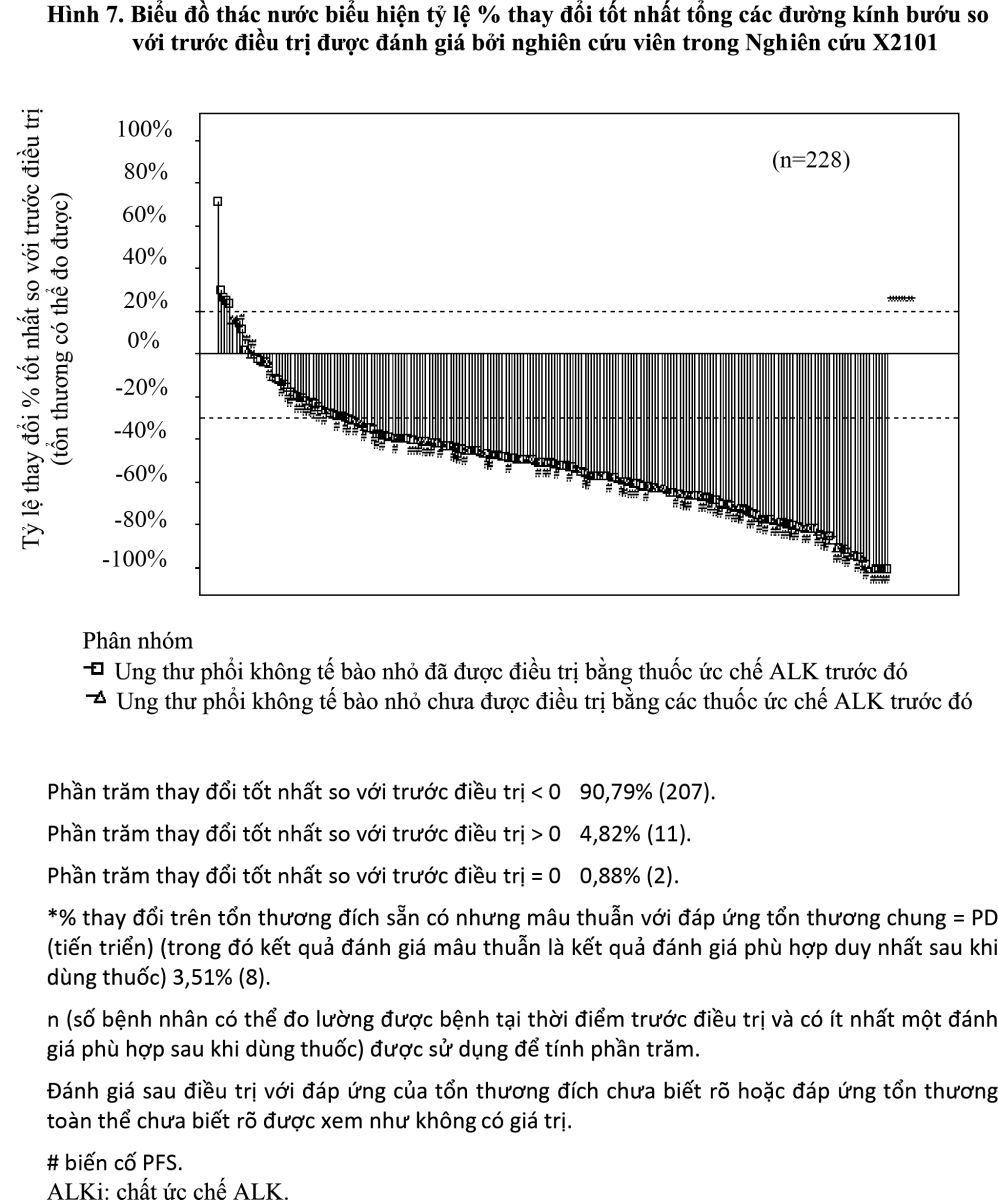
Trong Nghiên cứu X2101, trên 246 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK dùng liều 750 mg, tỷ lệ đáp ứng toàn bộ (ORR) theo đánh giá của Nghiên cứu viên là 61,8% (khoảng tin cậy 95%: 55,4; 67,9). Thời gian đáp ứng trung vị trên bệnh nhân có đáp ứng là 9,7 tháng (khoảng tin cậy 95%: 8,3; 11,4). Thời gian trung vị đến khi có đáp ứng bướu khách quan đầu tiên, sau đó được khẳng đinh, là 6,1 tuần (từ 3,0 đến 42,1). Thời gian sống thêm không bệnh tiến triển (PFS) là 9,0 tháng (khoảng tin cậy 95%: 6,9; 11,0). Bệnh nhân có đáp ứng với Spexib bất kể trước đó họ đã dùng thuốc ức chế ALK hay không theo số liệu Bảng 5. Dữ liệu chính về hiệu quả của thuốc từ 3 thử nghiệm lâm sàng được tóm tắt trong Bảng 5.
- xem Bảng 5.
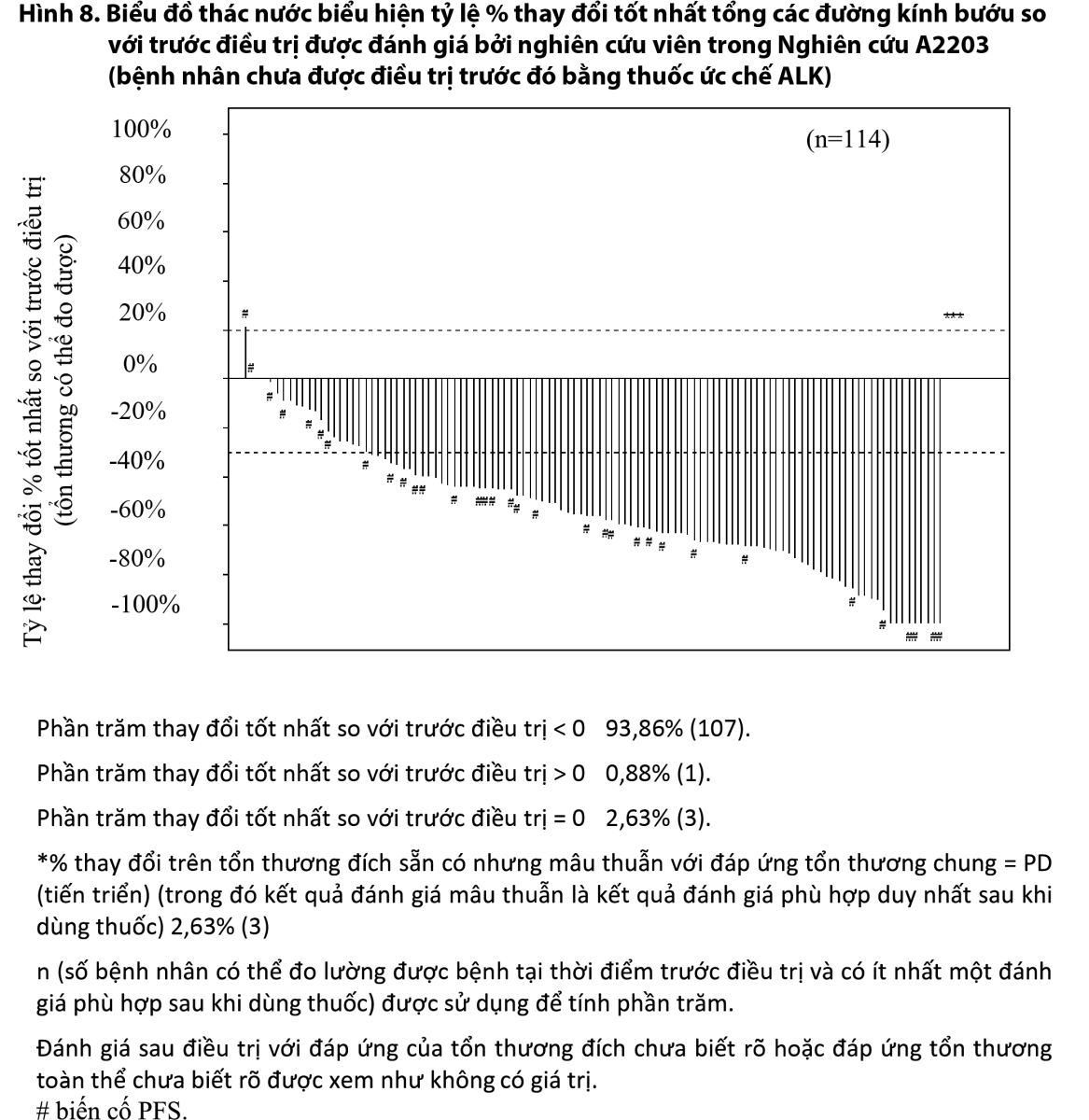
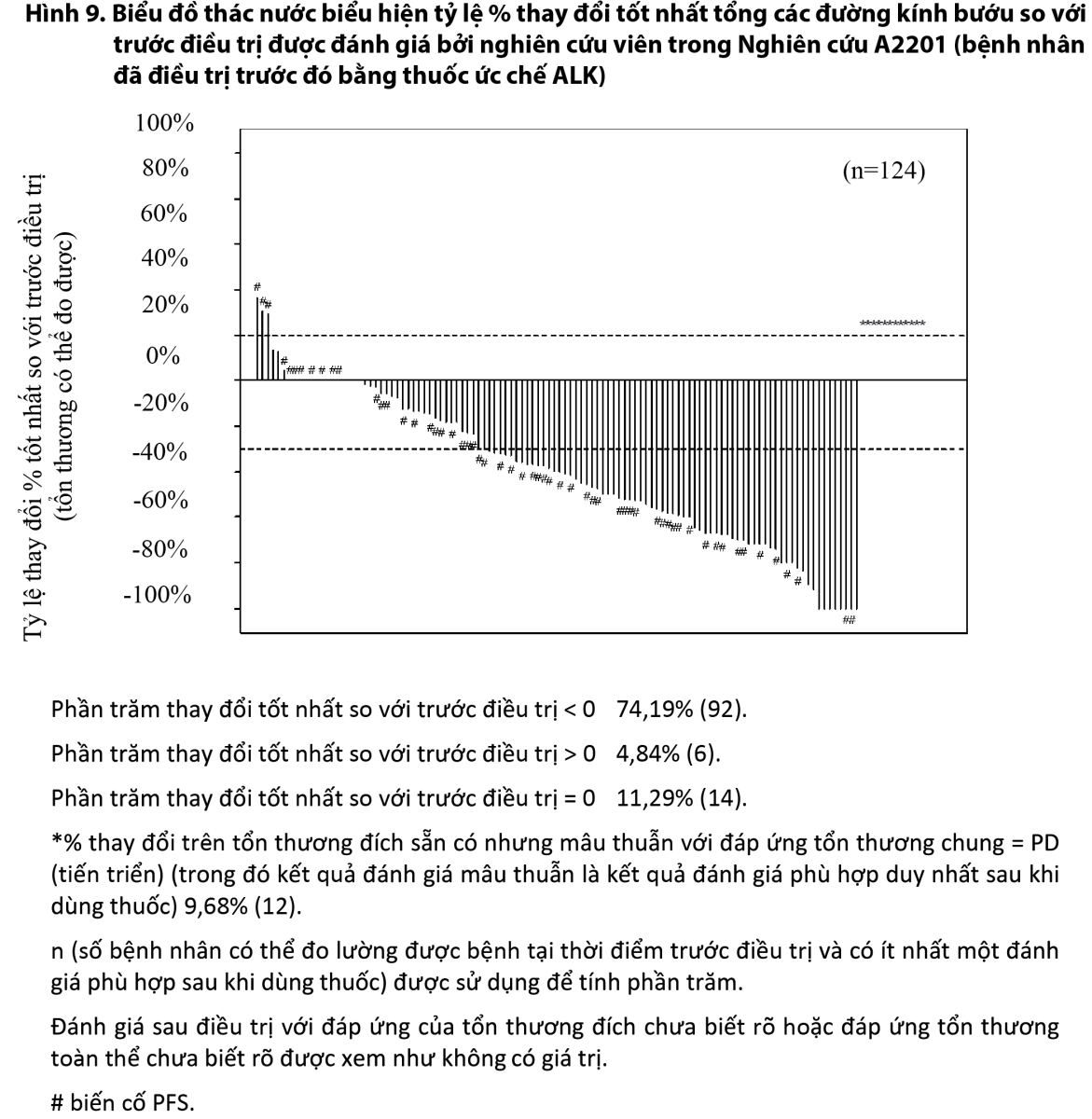
Biểu đồ thác nước hiển thị mức độ giảm tối đa tổng các đường kính lớn nhất của bướu so với trước khi điều trị trong cả 3 nghiên cứu cho thấy phần lớn bệnh nhân được điều trị bằng Spexib có sự giảm tổng khối bướu (Hình 7, Hình 8, Hình 9).
- xem Hình 7, Hình 8, Hình 9.
Đường cong Kaplan-Meier về thời gian sống thêm không bệnh tiến triển trong 3 nghiên cứu được trình bày trong Hình 10, Hình 11, và Hình 12.
- xem Hình 10, Hình 11, Hình 12.
Bệnh nhân có di căn não
Trong nghiên cứu X2101, A2203 và A2201, di căn não đã được ghi nhận tương ứng trên 50,0%, 40,3% và 71,4% bệnh nhân.
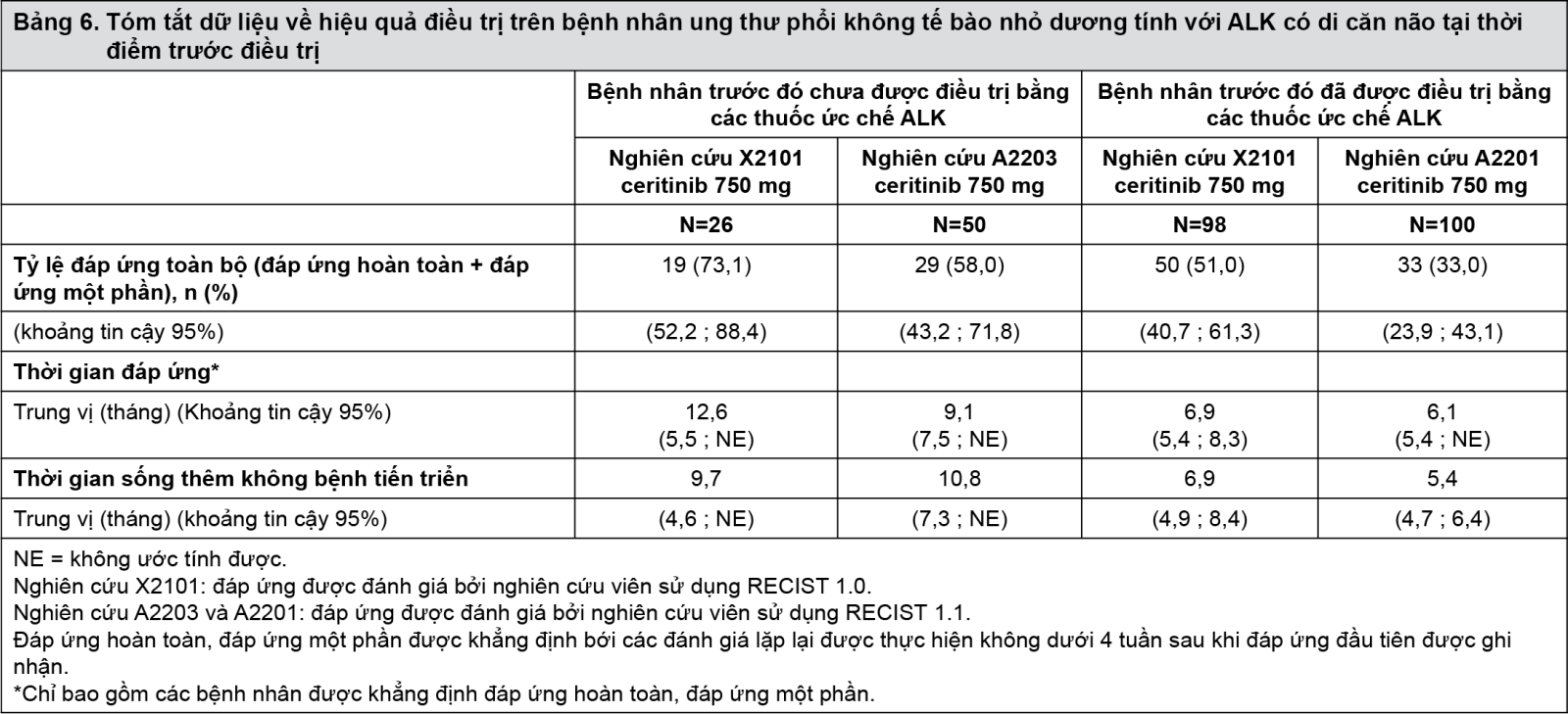
Các dữ liệu đánh giá hiệu quả chính trên bệnh nhân có di căn não trước điều trị trong 3 thử nghiệm lâm sàng được tóm tắt trong Bảng 6.
- xem Bảng 6.
Trong nghiên cứu X2101, có 14 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK có khối u di căn não có thể đo được tại thời điểm ban đầu theo đánh giá của nghiên cứu viên trong nhóm dùng mức liều 750 mg. Tỷ lệ đáp ứng nội sọ toàn bộ (OIRR) được đánh giá bởi các nghiên cứu viên là 50,0% (khoảng tin cậy 95%: 23,0; 77,0), bao gồm 2 bệnh nhân có đáp ứng hoàn toàn trong não và 5 bệnh nhân được khẳng định có đáp ứng một phần trong não; ngoài ra 3 bệnh nhân có bệnh ổn định (SD).
Trong nghiên cứu A2203, 10 trong số 124 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK có di căn não tại thời điểm ban đầu được đánh giá là tổn thương đích theo các nghiên cứu viên. Trên các bệnh nhân này, tỷ lệ đáp ứng nội sọ toàn bộ dựa trên đánh giá của nghiên cứu viên là 20,0% (khoảng tin cậy 90%: 2,5; 55,6), bao gồm 2 bệnh nhân được khẳng định có đáp ứng một phần trong não.
Trong nghiên cứu A2201, 20 trong số 140 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK có di căn não tại thời điểm ban đầu được xem như tổn thương đích theo đánh giá của nghiên cứu viên. Trong số các bệnh nhân này, tỷ lệ đáp ứng nội sọ toàn bộ dựa trên đánh giá của nghiên cứu viên là 35,0% (khoảng tin cậy 95%: 15,4; 59,2), bao gồm 2 bệnh nhân được khẳng định có đáp ứng hoàn toàn trong não và 5 bệnh nhân được khẳng định có đáp ứng một phần trong não.
Ngoài ra, đối với các bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK có tổn thương não có thể đo được tại thời điểm ban đầu chưa được xạ trị, Spexib tạo đáp ứng trong não tương tự hoặc nhiều hơn đáp ứng bướu toàn thân trên phần lớn các bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK đã hoặc chưa từng điều trị bằng thuốc ức chế ALK trước đó.
Trong nghiên cứu X2101, 41 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK tham gia vào nghiên cứu có di căn não chưa được xạ trị (30 bệnh nhân trước đó đã được điều trị bằng các thuốc ức chế ALK và 11 bệnh nhân chưa được điều trị bằng các thuốc ức chế ALK), 4 trong số các bệnh nhân này có tổn thương não đo được tại thời điểm ban đầu (3 bệnh nhân trước đó được điều trị bằng một thuốc ức chế ALK, 1 bệnh nhân trước đó chưa được điều trị bằng thuốc ức chế ALK). Tất cả 4 bệnh nhân (100%) có tổn thương não có thể đo được tại thời điểm ban đầu chưa được xạ trị có đáp ứng bướu tương tự hoặc nhiều hơn đáp ứng bướu toàn thân bao gồm 2 bệnh nhân có di căn não đáp ứng hoàn toàn (2 bệnh nhân trước đó đã được điều trị bằng một thuốc ức chế ALK và 1 bệnh nhân trước đó chưa được điều trị bằng thuốc ức chế ALK). Ngoài 2 bệnh nhân có đáp ứng hoàn toàn, còn có 1 bệnh nhân đáp ứng một phần và 1 trường hợp bệnh ổn định.
Trong nghiên cứu A2203, 23 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK tham gia vào nghiên cứu có di căn não chưa được xạ trị, 6 trong số bệnh nhân này có tổn thương não có thể đo được tại thời điểm ban đầu. Cả 6 bệnh nhân (100%) có đáp ứng trong não tương tự với đáp ứng bướu toàn thân. Có 2 bệnh nhân đáp ứng một phần, 3 bệnh ổn định và 1 bệnh nhân “chưa rõ”.
Trong nghiên cứu A2201, 28 bệnh nhân ung thư phổi không tế bào nhỏ dương tính với ALK tham gia vào nghiên cứu có di căn não chưa được xạ trị, 6 trong số bệnh nhân này có tổn thương não có thể đo được tại thời điểm ban đầu. 4 trong 6 bệnh nhân (66,7%) có đáp ứng trong não tương tự hoặc nhiều hơn đáp ứng bướu toàn thân bao gồm 2 bệnh nhân có đáp ứng di căn não hoàn toàn. Ngoài 2 bệnh nhân đáp ứng hoàn toàn, có 2 bệnh nhân đáp ứng một phần và 2 bệnh nhân bệnh ổn định.
ĐẶC TÍNH DƯỢC ĐỘNG HỌC
Hấp thu
Nồng độ đỉnh trong huyết tương (Cmax) của ceretinib đạt được khoảng 4 đến 6 giờ sau khi uống. Mức độ hấp thu đường uống ước tính ≥ 25% dựa trên tỷ lệ phần trăm chất chuyển hóa trong phân. Sinh khả dụng tuyệt đối của ceritinib chưa được xác định.
Nồng độ ceritinib trong tuần hoàn tăng lên khi dùng thuốc cùng với thức ăn. Diện tích dưới đường cong của ceritinib cao hơn khoảng 58% và 73% tương ứng (Cmax cao hơn khoảng 43% và 41%) khi dùng cùng bữa ăn ít chất béo và bữa ăn giàu chất béo.
Trong một nghiên cứu lâm sàng so sánh Spexib 450 mg hoặc 600 mg mỗi ngày dùng với thức ăn (khoảng 100 đến 500 calo và 1,5 đến 15 gram chất béo) với liều 750 mg mỗi ngày trong điều kiện nhịn ăn, phơi nhiễm hệ thống được quan sát ở trạng thái ổn định với nhóm dùng liều 450 mg với thức ăn (N=36) tương tự với nhóm nhịn ăn dùng liều 750 mg (N=31), chỉ có tăng nhẹ AUC (khoảng tin cậy (CI) 90%) dưới 4% (-13%, 24%) và Cmax (khoảng tin cậy (CI) 90%) dưới 3% (-14%, 22%). Ngược lại, AUC (khoảng tin cậy (CI) 90%) và Cmax (khoảng tin cậy (CI) 90%) cho nhóm dùng liều 600 mg với thức ăn (N=30) tăng tương ứng 24% (3%, 49%) và 25% (4%, 49%), so với nhóm nhịn ăn dùng liều 750 mg. Liều 600 mg hoặc cao hơn mỗi ngày dùng với thức ăn sẽ dẫn đến phơi nhiễm hệ thống cao hơn nhóm nhịn ăn dùng liều 750 mg và có thể làm tăng các phản ứng bất lợi của thuốc.
Sau khi bệnh nhân uống liều đơn ceritinib, lượng ceritinib trong huyết tương, thể hiện qua giá trị Cmax và AUClast, tăng tỷ lệ với liều trong khoảng liều từ 50 đến 750 mg. Trái với dữ liệu trên liều đơn, nồng độ trước khi dùng thuốc (Cmin) sau khi dùng liều lặp lại hàng ngày dường như tăng cao hơn so với mức độ tăng liều.
Phân bố
In vitro, ceritinib gắn với protein huyết tương người khoảng 97% trên, không phụ thuộc vào nồng độ trong khoảng từ 50 ng/mL đến 10.000 ng/mL. So với trong huyết tương, ceritinib cũng phân bố nhiều hơn trong tế bào hồng cầu, với tỷ lệ trung bình trong máu so với huyết tương trên in vitro là 1,35. Các nghiên cứu in vitro cho thấy ceritinib là cơ chất của P-glycoprotein (P-gp) nhưng không là cơ chất của protein kháng ung thư vú (BCRP) hoặc protein 2 đa kháng (MRP2). Khả năng thấm thụ động in vitro qua màng tế bào của ceritinib thấp.
Trên chuột cống, ceritinib qua được hàng rào máu não không bị tổn thương với tỷ số trong não so với trong máu (AUCinf) khoảng 15%. Chưa có các dữ liệu liên quan đến tỷ lệ thuốc trong não so với trong máu người.
Chuyển dạng sinh học/chuyển hóa
Các nghiên cứu in vitro cho thấy CYP3A là enzym chính tham gia vào quá trình chuyển hóa thải trừ của ceritinib.
Sau khi uống liều đơn 750 mg ceritinib gắn phóng xạ, ceritinib là thành phần chính lưu hành trong huyết tương người. Có tất cả 11 chất chuyển hóa đã được tìm thấy trong huyết tương ở nồng độ thấp với mức độ phân bố trung bình của mỗi chất chuyển hóa cho hoạt tính phóng xạ AUC ≤ 2,3%. Con đường chuyển hóa sinh học chủ yếu được xác định trên người khỏe mạnh bao gồm mono oxy hóa, O-dealkyl hóa và N-formyl hóa. Con đường chuyển hóa sinh học thứ cấp liên quan đến các chất chuyển hóa sinh học chủ yếu bao gồm quá trình glucuronid hóa và dehydrogen hóa. Gắn nhóm thiol vào ceritinib O-dealkyl hóa cũng đã được ghi nhận.
Thải trừ
Sau khi uống liều đơn ceritinib, trung bình nhân thời gian bán thải trong huyết tương (T1/2) của ceritinib dao động từ 31 đến 41 giờ ở bệnh nhân dùng liều từ 400 mg đến 750 mg. Liều uống hàng ngày của ceritinib đạt trạng thái ổn định sau khoảng 15 ngày và duy trì ổn định sau đó, với tỷ số tích lũy là 6,2 sau 3 tuần dùng liều hàng ngày. Thanh thải trung bình (CL/F) ở trạng thái ổn định của ceritinib thấp hơn (33,2 L/giờ) sau khi dùng liều uống hàng ngày 750 mg so với dùng liều đơn 750 mg (88,5 L/giờ) cho thấy dược động học của ceritinib không tuyến tính theo thời gian.
Con đường thải trừ chính của ceritinib và các chất chuyển hóa của nó là qua phân. Trung bình 68% liều dùng của ceritinib được tìm thấy trong phân dưới dạng không biến đổi. Chỉ 1,3% liều dùng được tìm thấy trong nước tiểu.
Nhóm bệnh nhân đặc biệt
Bệnh nhân suy giảm chức năng gan
Ceritinib chưa được nghiên cứu trên bệnh nhân suy giảm chức năng gan. Tuy nhiên dựa trên dữ liệu hiện có, ceritinib được thải trừ chủ yếu qua gan. Do đó, suy giảm chức năng gan có thể làm tăng nồng độ ceritinib trong huyết tương.
Dựa trên phân tích dược động học quần thể trên 140 bệnh nhân có suy giảm chức năng gan nhẹ (bilirubin toàn phần ≤ giới hạn trên của mức bình thường và AST > giới hạn trên của giá trị bình thường hoặc bilirubin toàn phần >1,0 đến 1,5 lần giới hạn trên của giá trị bình thường, không phụ thuộc vào giá trị AST) và 832 bệnh nhân có chức năng gan bình thường (bilirubin toàn phần ≤ giới hạn trên của giá trị bình thường và AST ≤ giới hạn trên của giá trị bình thường), nồng độ ceritinib trên bệnh nhân suy giảm chức năng gan nhẹ tương tự trên bệnh nhân có chức năng gan bình thường. Không khuyến cáo hiệu chỉnh liều trên bệnh nhân suy giảm chức năng gan nhẹ dựa trên các kết quả phân tích dược động học quần thể. Dược động học của ceritinib chưa được nghiên cứu trên bệnh nhân suy gan trung bình đến nặng. Liều khuyến cáo chưa được xác định trên bệnh nhân suy gan trung bình đến nặng.
Bệnh nhân suy giảm chức năng thận
Ceritinib chưa được nghiên cứu trên bệnh nhân suy giảm chức năng thận. Tuy nhiên, dựa trên các dữ liệu hiện có, lượng ceritinib thải trừ qua thận không đáng kể (1,3% liều đơn).
Dựa trên phân tích dược động học quần thể từ 345 bệnh nhân suy thận nhẹ (thanh thải creatinin 60 đến < 90mL/phút), 82 bệnh nhân suy thận ở mức độ trung bình (thanh thải creatinin 30 đến < 60mL/phút) và 546 bệnh nhân có chức năng thận bình thường (≥ 90mL/phút), nồng độ ceritinib trên bệnh nhân suy thận nhẹ và trung bình tương tự trên bệnh nhân có chức năng thận bình thường cho thấy không cần thiết phải hiệu chỉnh liều trên bệnh nhân suy thận nhẹ đến trung bình. Bệnh nhân suy thận nặng (thanh thải creatinin < 30mL/phút) không được thu nhận trong thử nghiệm lâm sàng.
Ảnh hưởng của tuổi, giới và chủng tộc
Phân tích dược động học quần thể cho thấy tuổi, giới và chủng tộc không ảnh hưởng có ý nghĩa lâm sàng đến nồng độ ceritinib.
Điện sinh lý tim
Khả năng kéo dài khoảng QT của ceritinib được đánh giá trong 7 thử nghiệm lâm sàng với Spexib. Điện tâm đồ hàng loạt được thu thập sau khi dùng liều đơn và ở trạng thái ổn định để đánh giá ảnh hưởng của ceretinib trên khoảng QT. Một phân tích trung tâm trên dữ liệu điện tâm đồ đã cho thấy khoảng QTc >500 ms xuất hiện mới trên 12 bệnh nhân (1,3%). Có 58 bệnh nhân (6,3%) có khoảng QTc tăng so với ban đầu >60 ms. Một phân tích xu hướng trung tâm của dữ liệu khoảng QTc tại các nồng độ trung bình ở trạng thái ổn định từ một nghiên cứu lâm sàng toàn cầu pha 3 (Nghiên cứu A2301) đã chứng minh giới hạn trên của khoảng tin cậy 90% 2 phía của khoảng QTc là 15,3 ms đối với ceritinib dùng liều 750 mg. Một phân tích dược động học/dược lực học cho thấy ceritinib làm tăng khoảng QTc phụ thuộc nồng độ (xem mục Cảnh báo).
THÔNG TIN TIỀN LÂM SÀNG
Dược lý an toàn
Nghiên cứu dược lý an toàn cho thấy ceritinib không gây ảnh hưởng đến các chức năng sinh tồn của hệ hô hấp và thần kinh trung ương. Dữ liệu in vitro cho thấy giá trị ức chế IC50 của ceritinib trên kênh kali hERG là 0,4 mM ở nhiệt độ từ 33oC đến 35oC (gần với nhiệt độ cơ thể). Một nghiên cứu in vivo từ xa trên khỉ cho thấy có sự kéo dài nhẹ khoảng QT trên 1 trong số 4 con sau khi dùng ceritinib liều cao nhất. Nghiên cứu điện tâm đồ trên khỉ sau 4 hoặc 13 tuần dùng ceritinib không cho thấy khoảng QT bị kéo dài cũng như các bất thường trên điện tâm đồ.
Độc tính trên gen
Thử nghiệm Ames cho thấy ceritinib không gây đột biến gen và thử nghiệm sai lệch nhiễm sắc thể trên tế bào lympho ngoại vi máu người được nuôi cấy không cho thấy thuốc có khả năng gây sai lệch nhiễm sắc thể. Thử nghiệm vi nhân sử dụng tế bào lympho máu người cho kết quả âm tính. Thử nghiệm vi nhân in vivo trên chuột cống không cho thấy phản ứng bất lợi của thuốc trên tủy xương sau khi cho chuột uống thuốc.
Khả năng gây ung thư và đột biến gen
Nghiên cứu về khả năng gây ung thư chưa được thực hiện với ceritinib.
Có thai/khả năng sinh sản
Nghiên cứu độc tính trên sinh sản (như nghiên cứu trên sự phát triển bào thai – thai nhi) trên chuột cống và thỏ có thai cho thấy thuốc không gây độc với bào thai hoặc gây dị tật sau khi dùng ceritinib trong giai đoạn hình thành các cơ quan. Tuy nhiên, nồng độ thuốc trong huyết tương mẹ thấp hơn so với ghi nhận được ở liều được khuyến cáo 750 mg trong các thử nghiệm lâm sàng. Các nghiên cứu tiền lâm sàng chính thức về ảnh hưởng của ceritinib trên khả năng sinh sản chưa được thực hiện.
Nghiên cứu độc tính liều lặp lại
Độc tính chủ yếu liên quan đến ceritinib trên chuột cống và khỉ là viêm đường mật ngoài gan có kèm theo tăng số lượng bạch cầu trung tính ngoại vi. Viêm hỗn hợp tế bào/bạch cầu trung tính đường mật ngoài gan lan rộng đến tuyến tụy và/hoặc tá tràng với liều cao hơn. Độc tính trên đường tiêu hóa đã được ghi nhận trên cả hai loài là giảm cân, giảm tiêu thụ thức ăn, nôn (trên khỉ), tiêu chảy và ở liều cao có tổn thương mô bệnh học bao gồm ăn mòn, viêm niêm mạc và các đại thực bào có bọt trong các khe niêm mạc tá tràng và lớp dưới niêm mạc. Gan cả hai loài cũng bị ảnh hưởng, nhưng chỉ khi nghiên cứu ở mức liều cao nhất bao gồm tăng tối thiểu các enzym transaminase gan trên một số động vật và xuất hiện không bào của biểu mô đường mật trong gan. Đại thực bào phế nang có bọt (khẳng định có hiện tượng phospholipidosis) đã được ghi nhận trên phổi chuột cống nhưng không được ghi nhận trên khỉ, và các hạch bạch huyết của chuột và khỉ có đại thực bào lắng đọng. Tổn thương cơ quan đích được ghi nhận phục hồi một phần hoặc hoàn toàn.
Chỉ định/Công dụng
Spexib được chỉ định điều trị cho bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC) tiến xa tại chỗ hoặc di căn, dương tính với ALK (anaplastic lymphoma kinase).
Liều lượng & Cách dùng
Nhóm bệnh nhân đích nói chung
Liều được khuyến cáo của Spexib là 750 mg dùng đường uống một lần mỗi ngày lúc đói (ít nhất một giờ trước bữa ăn hoặc hai giờ sau bữa ăn) tại cùng một thời điểm mỗi ngày.
Liều tối đa được khuyến cáo là 750 mg/ngày.
Tiếp tục điều trị nếu thuốc còn đem lại lợi ích lâm sàng cho bệnh nhân.
Thay đổi liều
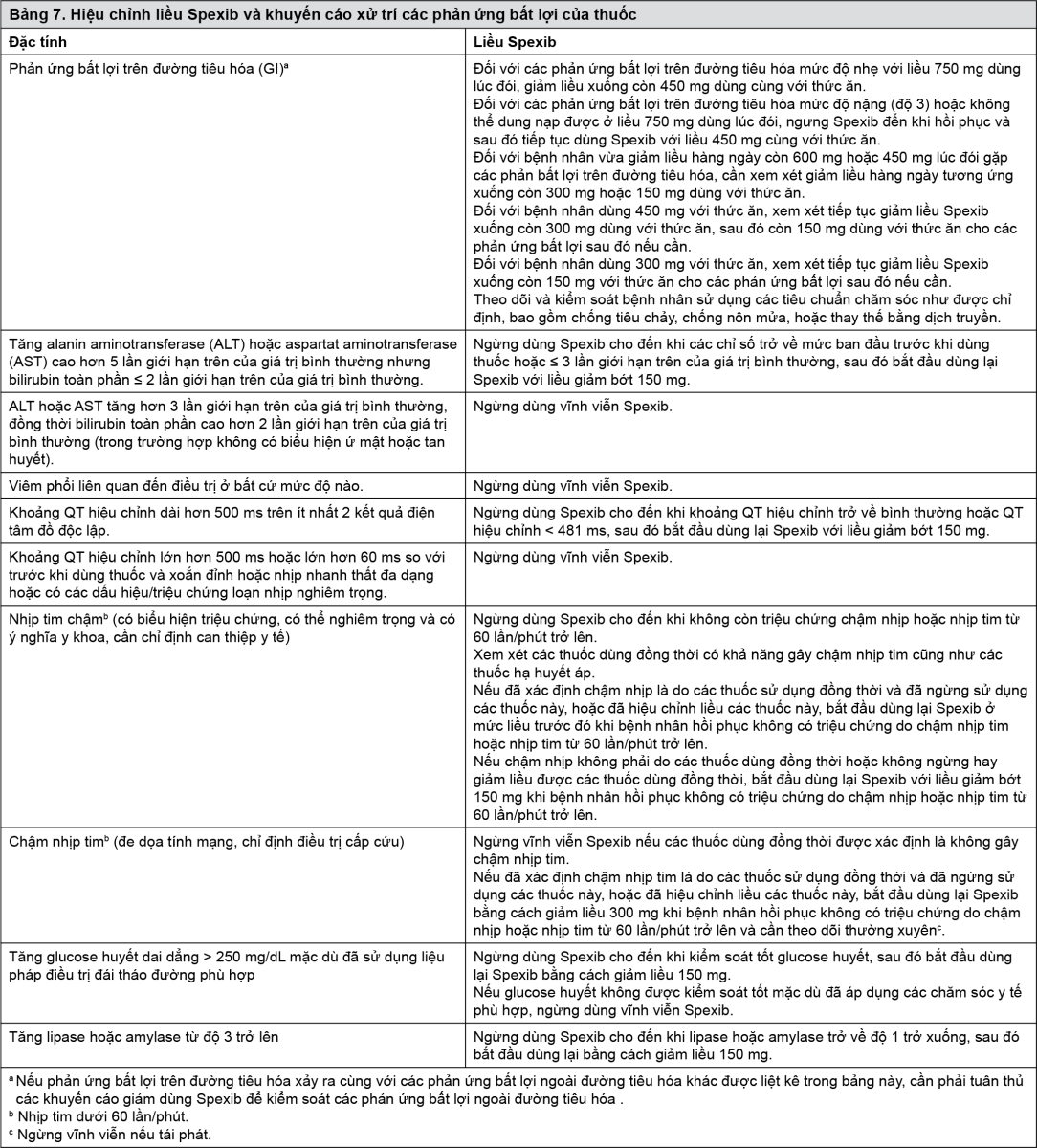
Dựa trên độ an toàn và khả năng dung nạp của bệnh nhân, có thể cần tạm ngừng và/hoặc giảm liều Spexib. Nếu cần giảm liều do một phản ứng bất lợi nào của thuốc không được nêu trong bảng 1, nên giảm liều dùng hàng ngày Spexib dần từng bước mỗi lần 150 mg. Cân nhắc phát hiện sớm và xử trí các phản ứng bất lợi của thuốc bằng các biện pháp chăm sóc hỗ trợ chuẩn.
Nên ngừng dùng Spexib nếu bệnh nhân không dung nạp được liều 300 mg/ngày lúc đói hoặc liều 150 mg/ngày khi dùng cùng thức ăn.
Phản ứng bất lợi trên đường tiêu hóa
Đối với bệnh nhân uống 750 mg/ngày khi đói gặp các phản ứng bất lợi trên đường tiêu hóa, giảm liều hàng ngày xuống còn 450 mg dùng với thức ăn để giảm kích ứng tại chỗ đường tiêu hoá trong khi vẫn duy trì dùng Spexib (xem phần Dược lý). Các khuyến cáo điều chỉnh liều bổ sung để kiểm soát các phản ứng bất lợi trên đường tiêu hóa được cung cấp trong Bảng 7.
Nếu phản ứng bất lợi trên đường tiêu hóa xảy ra cùng với các phản ứng bất lợi ngoài đường tiêu hóa khác được liệt kê trong Bảng 7, cần phải tuân thủ các khuyến cáo giảm liều dùng Spexib để kiểm soát các phản ứng bất lợi cụ thể ngoài đường tiêu hóa (Bảng 7).
Bảng 7 tóm tắt các khuyến cáo về tạm ngừng, giảm liều hoặc ngừng vĩnh viễn Spexib trong quá trình xử trí một số phản ứng bất lợi (ADR) cụ thể của thuốc.
- xem Bảng 7.
Trong quá trình điều trị bằng Spexib, tránh dùng các thuốc ức chế mạnh CYP3A (xem mục Tương tác). Nếu bắt buộc phải sử dụng đồng thời với thuốc ức chế mạnh CYP3A, giảm liều còn khoảng 1/3 mức liều Spexib, làm tròn đến bội số gần nhất của mức liều 150 mg. Sau khi ngừng dùng thuốc ức chế mạnh CYP3A, quay trở lại dùng liều Spexib như trước khi bắt đầu dùng thuốc ức chế mạnh CYP3A.
Nhóm bệnh nhân đặc biệt
Bệnh nhân suy giảm chức năng thận
Không cần hiệu chỉnh liều cho bệnh nhân suy thận ở mức độ nhẹ đến trung bình. Nên thận trọng khi dùng thuốc cho bệnh nhân suy thận nặng do chưa có kinh nghiệm sử dụng Spexib cho quần thể bệnh nhân này (xem mục Dược lý).
Bệnh nhân suy giảm chức năng gan
Không cần hiệu chỉnh liều cho bệnh nhân suy giảm chức năng gan nhẹ. Nên thận trọng khi dùng thuốc cho bệnh nhân suy giảm chức năng gan ở mức độ trung bình đến nặng (xem mục Dược lý).
Bệnh nhi
Độ an toàn và hiệu quả của Spexib chưa được thiết lập trên đối tượng bệnh nhi.
Bệnh nhân cao tuổi (≥65 tuổi)
Các dữ liệu hạn chế về độ an toàn và hiệu quả của Spexib trên bệnh nhân từ 65 tuổi trở lên cho thấy không cần hiệu chỉnh liều trên bệnh nhân cao tuổi (xem mục Dược lý).
Cách dùng
Nên uống Spexib 1 lần/ngày lúc đói (ít nhất một giờ trước bữa ăn hoặc hai giờ sau bữa ăn vào cùng một thời điểm hàng ngày (xem mục Dược lý). Nên nuốt cả viên nang Spexib với nước. Không nên nhai hoặc bẻ viên nang.
Nếu bệnh nhân gặp các phản ứng bất lợi trên đường tiêu hóa cần xem xét giảm liều Spexib và dùng cùng với thức ăn (xem phần Liều lượng và Cách dùng).
Nếu quên uống thuốc, bệnh nhân không nên dùng liều đó mà dùng liều quy định tiếp theo.
Cảnh báo
Độc tính trên gan
Độc tính trên gan xảy ra với tỷ lệ 1,1% bệnh nhân điều trị bằng Spexib trong các nghiên cứu lâm sàng. Tăng ALT ở mức độ 3 hoặc 4 được ghi nhận trên 25% bệnh nhân dùng Spexib. ALT tăng hơn 3 lần giới hạn trên của giá trị bình thường kèm theo bilirubin toàn phần tăng gấp 2 lần giới hạn trên của giá trị bình thường, trong khi phosphatase kiềm không tăng, được ghi nhận trong số dưới 1% bệnh nhân trong các nghiên cứu lâm sàng. Đa số các trường hợp có thể kiểm soát được bằng cách ngừng và/hoặc giảm liều. Một vài trường hợp phải ngừng dùng Spexib.
Theo dõi các xét nghiệm chức năng gan (bao gồm ALT, AST và bilirubin toàn phần) trước khi bắt đầu điều trị và hàng tháng sau đó. Ở bệnh nhân có tăng transaminase, nên theo dõi transaminase gan và bilirubin toàn phần thường xuyên hơn theo chỉ định lâm sàng (xem mục Liều lượng và Cách dùng và mục Tác dụng ngoại ý).
Bệnh phổi mô kẽ/Viêm phổi
Bệnh phổi mô kẽ nghiêm trọng, đe dọa tính mạng hoặc gây tử vong hoặc bệnh viêm phổi đã được ghi nhận trên bệnh nhân điều trị bằng Spexib trong các nghiên cứu lâm sàng. Hầu hết các trường hợp nặng/đe dọa tính mạng này đã cải thiện hoặc đã khỏi sau khi tạm ngừng dùng Spexib.
Theo dõi các bệnh nhân có triệu chứng biểu hiện của viêm phổi. Loại trừ các nguyên nhân khác gây viêm phổi và ngừng sử dụng vĩnh viễn Spexib cho bệnh nhân được chẩn đoán viêm phổi do thuốc (xem mục Liều lượng và Cách dùng và mục Tác dụng ngoại ý).
Kéo dài khoảng QT
Kéo dài khoảng QTc trên điện tâm đồ đã được ghi nhận trong các thử nghiệm lâm sàng trên bệnh nhân được điều trị bằng Spexib. Kéo dài khoảng QTc có thể dẫn đến làm tăng nguy cơ loạn nhịp nhanh thất (như xoắn đỉnh) hoặc đột tử. Một phân tích trung tâm trên dữ liệu điện tâm đồ cho thấy khoảng QTc >500 ms xuất hiện mới trên 12 bệnh nhân (1,3%), trong đó 6 người có khoảng QTc tăng >450 ms so với ban đầu. Có 58 bệnh nhân (6,3%) có khoảng QTc tăng >60 ms so với ban đầu. Một phân tích dược động học/dược lực học cho thấy ceritinib gây tăng khoảng QTc phụ thuộc nồng độ.
Tránh sử dụng Spexib cho bệnh nhân có hội chứng kéo dài khoảng QT bẩm sinh. Khuyến cáo theo dõi thường kỳ điện tâm đồ và điện giải (như kali) cho bệnh nhân bị suy tim sung huyết, loạn nhịp chậm hoặc các bất thường về điện giải và trên bệnh nhân đang dùng các thuốc làm kéo dài khoảng QT. Điều chỉnh điện giải theo chỉ định lâm sàng trong trường hợp nôn, tiêu chảy, mất nước hoặc suy giảm chức năng thận. Ngừng dùng vĩnh viễn Spexib cho bệnh nhân có khoảng QTc trên 500 ms hoặc dài hơn 60 ms so với trước khi dùng thuốc và có xoắn đỉnh hoặc nhịp nhanh thất đa dạng hoặc các dấu hiệu/triệu chứng của loạn nhịp nghiêm trọng. Ngừng dùng Spexib cho bệnh nhân có khoảng QTc >500 ms trên ít nhất 2 kết quả điện tâm đồ riêng rẽ cho đến khi điện tâm đồ trở về bình thường hoặc khoảng QTc dưới 481 ms, sau đó bắt đầu dùng lại Spexib bằng cách giảm liều 150 mg (xem mục Liều lượng và Cách dùng, mục Tác dụng ngoại ý và mục Dược lý).
Chậm nhịp tim
Các trường hợp chậm nhịp tim không có biểu hiện triệu chứng đã được ghi nhận trên bệnh nhân dùng Spexib trong các nghiên cứu lâm sàng.
Tránh dùng phối hợp Spexib với các thuốc khác gây chậm nhịp tim (như thuốc chẹn beta, thuốc chẹn kênh canxi không phải là dẫn chất dihydropyridin, clonidin và digoxin) trong khả năng có thể. Theo dõi thường xuyên nhịp tim và huyết áp. Trong trường hợp nhịp chậm có biểu hiện triệu chứng nhưng không đe dọa tính mạng, ngừng dùng Spexib cho đến khi hồi phục chậm nhịp tim không còn biểu hiện triệu chứng hoặc nhịp tim từ 60 lần/phút trở lên, xem xét các thuốc dùng đồng thời và hiệu chỉnh liều Spexib nếu cần thiết. Ngừng dùng vĩnh viễn Spexib trong trường hợp chậm nhịp tim đe dọa tính mạng nếu chậm nhịp tim không phải do các thuốc dùng cùng gây ra. Tuy nhiên, nếu dùng Spexib cùng các thuốc đã được biết gây chậm nhịp tim hoặc hạ huyết áp, ngừng dùng Spexib cho đến khi hồi phục chậm nhịp tim không còn biểu hiện triệu chứng hoặc nhịp tim từ 60 lần/phút trở lên. Trong trường hợp có thể ngừng các thuốc dùng cùng hoặc hiệu chỉnh liều các thuốc này, bắt đầu dùng lại Spexib bằng cách giảm liều 300 mg cho đến khi hồi phục chậm nhịp tim không còn biểu hiện triệu chứng hoặc nhịp tim từ 60 lần/phút trở lên và theo dõi thường xuyên (xem mục Liều lượng và Cách dùng và mục Tác dụng ngoại ý).
Phản ứng bất lợi về đường tiêu hóa
Trong các nghiên cứu lâm sàng với Spexib, tiêu chảy, buồn nôn và nôn được ghi nhận rất phổ biến; 12,5% bệnh nhân được ghi nhận có tiêu chảy, buồn nôn hoặc nôn ở mức độ 3/4.
Theo dõi và quản lý bệnh nhân theo chương trình chăm sóc chuẩn, bao gồm chống tiêu chảy, chống nôn hoặc bù dịch theo chỉ định lâm sàng. Ngừng hoặc giảm liều dùng thuốc với thức ăn có thể được áp dụng trong trường hợp cần thiết (xem mục Liều lượng và Cách dùng và mục Tác dụng ngoại ý). Nếu xuất hiện nôn trong quá trình điều trị, bệnh nhân không nên dùng liều bổ sung nhưng nên tiếp tục dùng liều theo lịch trình tiếp theo.
Tăng glucose huyết
Biến cố tăng glucose huyết (tất cả các mức độ) đã được ghi nhận dưới 10% bệnh nhân điều trị bằng Spexib trong các thử nghiệm lâm sàng; 5,4% bệnh nhân được ghi nhận có biến cố ở mức độ 3/4. Nguy cơ tăng glucose huyết cao hơn trên bệnh nhân đái tháo đường và/hoặc sử dụng đồng thời các thuốc steroid.
Theo dõi glucose huyết lúc đói trước khi bắt đầu điều trị bằng Spexib và định kỳ sau đó theo chỉ định lâm sàng. Bắt đầu sử dụng hoặc tối ưu hóa các thuốc chống tăng glucose huyết theo chỉ định (xem mục Liều lượng và Cách dùng và mục Tác dụng ngoại ý).
Tăng lipase và/hoặc amylase
Tăng lipase và/hoặc amylase đã được ghi nhận trên bệnh nhân dùng Spexib trong các nghiên cứu lâm sàng.
Theo dõi nồng độ lipase và amylase trước khi bắt đầu điều trị bằng Spexib và định kỳ sau đó theo chỉ định lâm sàng (xem mục Liều lượng và Cách dùng và mục Tác dụng ngoại ý).
Ảnh hưởng của thuốc đối với công việc
Spexib ít ảnh hưởng đến khả năng lái xe và vận hành máy móc. Vì bệnh nhân có thể thấy mệt mỏi hoặc là rối loạn thị giác do đó cần thận trọng khi lái xe và vận hành máy móc trong suốt quá trình điều trị.
Quá Liều
Quá liều: Chưa có kinh nghiệm về quá liều Spexib được ghi nhận trên người.
Cách xử lý: Nên tiến hành các biện pháp điều trị hỗ trợ chung trong tất cả các trường hợp quá liều.
Chống chỉ định
Quá mẫn với ceritinib hoặc bất cứ thành phần nào khác của thuốc.
Sử dụng ở phụ nữ có thai và cho con bú
Phụ nữ có khả năng mang thai (và nếu các biện pháp tránh thai có thể được áp dụng)
Phụ nữ có khả năng mang thai nên được khuyến cáo sử dụng các biện pháp tránh thai hiệu quả cao trong khi dùng Spexib và 3 tháng sau khi ngừng điều trị.
Phụ nữ có thai
Chưa có các dữ liệu về sử dụng Spexib cho phụ nữ có thai. Các nghiên cứu về độc tính sinh sản (như nghiên cứu trên sự phát triển của bào thai-thai nhi) trên chuột cống và thỏ có thai cho thấy dùng ceritinib không gây độc với thai nhi hoặc không gây dị tật trong giai đoạn hình thành các cơ quan. Tuy nhiên, nồng độ thuốc trong huyết tương mẹ thấp hơn mức đã được ghi nhận khi dùng liều khuyến cáo là 750 mg trong các thử nghiệm lâm sàng. Nguy cơ trên người chưa được biết rõ. Không nên dùng Spexib cho phụ nữ có thai trừ khi lợi ích tiềm tàng của thuốc vượt trội nguy cơ trên thai nhi.
Phụ nữ đang cho con bú
Chưa rõ ceritinib có bài tiết vào sữa mẹ hay không. Do nhiều thuốc bài tiết qua sữa mẹ và do nguy cơ các phản ứng bất lợi nghiêm trọng trên trẻ sơ sinh, nên quyết định ngừng cho con bú hoặc ngừng sử dụng Spexib dựa trên mức độ quan trọng của Spexib đối với người mẹ.
Khả năng sinh sản
Chưa biết rõ về khả năng gây vô sinh của Spexib trên bệnh nhân nam và nữ.
Tương tác
Thuốc có thể gây tăng nồng độ ceritinib trong huyết tương
Trên đối tượng khỏe mạnh, sử dụng đồng thời liều đơn ceritinib 450 mg với ketoconazol (200 mg x 2 lần/ngày trong 14 ngày), một thuốc ức chế mạnh CYP3A/P-gp, làm tăng AUC của ceritinib 2,9 lần và tăng Cmax 1,2 lần so với dùng ceritinib đơn độc. AUC ở trạng thái ổn định của ceritinib với liều giảm khi dùng cùng ketoconazol 200 mg x 2 lần/ngày trong 14 ngày được mô phỏng tương tự AUC của ceritinib dùng đơn độc ở trạng thái ổn định. Nếu bắt buộc phải sử dụng cùng các thuốc ức chế mạnh CYP3A, bao gồm nhưng không giới hạn trong danh sách các thuốc sau: ritonavir, saquinavir, telithromycin, ketoconazol, itraconazol, voriconazol, posaconazol và nefazodon, giảm liều ceritinib đi khoảng 1/3, làm tròn đến bội số gần nhất của mức liều 150 mg. Sau khi ngừng dùng các thuốc ức chế mạnh CYP3A, dùng lại liều ceritinib như trước khi dùng các thuốc ức chế mạnh CYP3A.
Dựa trên các dữ liệu in vitro, ceritinib là cơ chất của chất vận chuyển qua màng P-glycoprotein (P-gp). Nếu dùng ceritinib cùng các thuốc ức chế P-gp có thể làm gia tăng nồng độ ceritinib. Thận trọng khi sử dụng đồng thời với các thuốc ức chế P-gp và theo dõi cẩn thận các phản ứng bất lợi của thuốc.
Các thuốc có thể làm giảm nồng độ ceritinib trong huyết tương
Trên đối tượng khỏe mạnh, sử dụng đồng thời ceritinib liều đơn với rifampicin (600 mg hàng ngày trong 14 ngày), một thuốc gây cảm ứng mạnh CYP3A/P-gp, làm giảm 70% AUC của ceritinib và 44% Cmax so với khi dùng ceritinib đơn độc. Sử dụng đồng thời ceritinib với các thuốc gây cảm ứng mạnh CYP3A/P-gp làm giảm nồng độ ceritinib trong huyết tương. Tránh sử dụng đồng thời với các thuốc gây cảm ứng mạnh CYP3A, bao gồm nhưng không giới hạn carbamazepin, phenobarbital, phenytoin, rifabutin, rifampin và St. John’s Wort (Hypericum perforatum). Thận trọng khi dùng đồng thời với các thuốc gây cảm ứng P-gp.
Các thuốc có nồng độ trong huyết tương thay đổi do ceritinib
Dựa trên các dữ liệu in vitro, ceritinib ức chế cạnh tranh quá trình chuyển hóa của midazolam, cơ chất của CYP3A, và diclofenac, cơ chất của CYP2C9. Tác dụng ức chế CYP3A phụ thuộc thời gian cũng được ghi nhận. Giá trị Cmax của ceritinib ở trạng thái ổn định ở mức liều được khuyến cáo trên lâm sàng là 750 mg/ngày có thể vượt quá giá trị Ki ức chế CYP3A và CYP2C9 cho thấy ceritinib có thể ức chế thanh thải của các thuốc khác được chuyển hóa bởi các enzym này ở nồng độ có ý nghĩa lâm sàng. Có thể cần thiết phải giảm liều khi dùng đồng thời với các thuốc chuyển hóa chủ yếu bởi CYP3A và CYP2C9. Tránh dùng đồng thời ceritinib với các cơ chất của CYP3A đã được biết có chỉ số điều trị hẹp (như astemizol, cisaprid, ciclosporin, ergotamin, fentanyl, pimozid, quinidin, tacrolimus, alfentanil và sirolimus) và cơ chất của CYP2C9 đã được biết có chỉ số điều trị hẹp (như phenytoin và warfarin).
Dựa trên các dữ liệu in vitro, ceritinib cũng ức chế CYP2A6 và CYP2E1 ở nồng độ liên quan trên lâm sàng. Do đó, ceritinib có thể làm tăng nồng độ trong huyết tương của các thuốc dùng cùng được chuyển hóa chủ yếu bởi các enzym này. Thận trọng khi dùng thuốc đồng thời với các cơ chất của CYP2A6 và CYP2E1 và theo dõi thận trọng các phản ứng bất lợi.
Các thuốc là cơ chất của các kênh vận chuyển
Dựa trên các dữ liệu in vitro, ceritinib không ức chế các bơm xuất bào không điển hình như BCRP, P-gp hoặc MRP2, các kênh vận chuyển tái thu hồi tại tế bào gan như OATP1B1 hay OATP1B3, kênh vận chuyển tái hấp thu anion hữu cơ tại thận OAT1 và OAT3 hoặc kênh vận chuyển tái hấp thu cation hữu cơ OCT1 hoặc OCT2 ở nồng độ có ý nghĩa lâm sàng. Do đó, không có khả năng xảy ra tương tác thuốc-thuốc có ý nghĩa lâm sàng do sự ức chế ceritinib qua trung gian các cơ chất của các kênh vận chuyển này.
Các chất ảnh hưởng đến pH dạ dày
Các thuốc làm giảm acid dạ dày (như các thuốc ức chế bơm proton, thuốc kháng H2, thuốc kháng acid) có thể làm thay đổi độ tan của ceritinib và làm giảm sinh khả dụng của nó vì ceritinib có độ hòa tan phụ thuộc vào pH và trở nên kém hòa tan khi pH tăng in vitro. Trong một nghiên cứu tương tác thuốc ở người khỏe mạnh (N=22), dùng kết hợp liều đơn 750 mg ceritinib và esomeprazole (thuốc ức chế bơm proton) liều 40 mg mỗi ngày trong 6 ngày làm giảm nồng độ ceritinib (AUCinf và Cmax giảm tương ứng 76% và 79%). Tuy nhiên, dùng kết hợp liều đơn 750 mg ceritinib với các thuốc ức chế bơm proton trong 6 ngày trong một phân nhóm bệnh nhân từ nghiên cứu X2101 cho thấy khả năng phơi nhiễm với ceritinib kém hơn so với nhóm người khỏe mạnh như AUC (khoảng tin cậy 90%) giảm 30% (0%, 52%) và Cmax (khoảng tin cậy 90%) giảm 25% (5%, 41%) và không quan sát thấy ảnh hưởng có ý nghĩa lâm sàng đến nồng độ ceritinib ở trạng thái ổn định sau khi dùng ceritinib một lần mỗi ngày.
Điều này tiếp tục được khẳng định bởi một phân tích phân nhóm dựa trên ba nghiên cứu lâm sàng (N >400) trong đó những bệnh nhân có dùng và không dùng thuốc ức chế bơm proton cho thấy tương đương về nồng độ ở trạng thái ổn định và hiệu quả và an toàn về mặt lâm sàng.
Tương tác thuốc-thức ăn/đồ uống
Sinh khả dụng của ceritinib tăng khi có mặt thức ăn phụ thuộc vào lượng chất béo trong bữa ăn (xem mục Dược lý). Nên dùng Spexib lúc đói. Không nên ăn ít nhất 2 giờ trước và 1 giờ sau khi dùng Spexib.
Nếu bệnh nhân gặp các phản ứng bất lợi trên đường tiêu hóa cần xem xét giảm liều Spexib dùng với thức ăn (xem phần Liều lượng và Cách dùng).
Bệnh nhân nên được hướng dẫn tránh dùng bưởi hoặc nước ép bưởi do chúng ức chế CYP3A ở thành ruột và có thể làm tăng sinh khả dụng của ceritinib.
Tác dụng ngoại ý
Tóm tắt dữ liệu về độ an toàn
Các phản ứng bất lợi của thuốc dưới đây phản ánh tình hình sử dụng Spexib trên 925 bệnh nhân ung thư phổi không tế bào nhỏ tiến xa dương tính với ALK được điều trị với liều khởi đầu uống 750 mg một lần mỗi ngày trong 7 nghiên cứu lâm sàng bao gồm hai nghiên cứu ngẫu nhiên, pha 3, có đối chứng chủ động.
Thời gian trung vị sử dụng Spexib là 44,9 tuần (dao động từ 0,1 đến 200,1 tuần). Giảm liều được ghi nhận trên 62,2% bệnh nhân và tạm ngừng liều được ghi nhận trên 74,8% bệnh nhân. Tỷ lệ các biến cố bất lợi dẫn đến phải ngừng dùng thuốc vĩnh viễn là 12,1%. Các biến cố bất lợi thường gặp nhất (>0,5%) dẫn đến phải ngừng dùng thuốc là viêm phổi (0,6%) và suy hô hấp (0,6%).
Các phản ứng bất lợi của thuốc với tần suất ≥10% là tiêu chảy, buồn nôn, nôn, các xét nghiệm gan bất thường, mệt mỏi, đau bụng, giảm cảm giác ăn ngon, giảm cân, táo bón, tăng creatinin máu, ban đỏ, thiếu máu và rối loạn thực quản.
Các phản ứng bất lợi độ 3/4 với tần suất ≥5% bao gồm các xét nghiệm chức năng gan bất thường, mệt mỏi, nôn, tăng glucose máu, buồn nôn và tiêu chảy.
Bảng tóm tắt các phản ứng bất lợi của thuốc từ các thử nghiệm lâm sàng
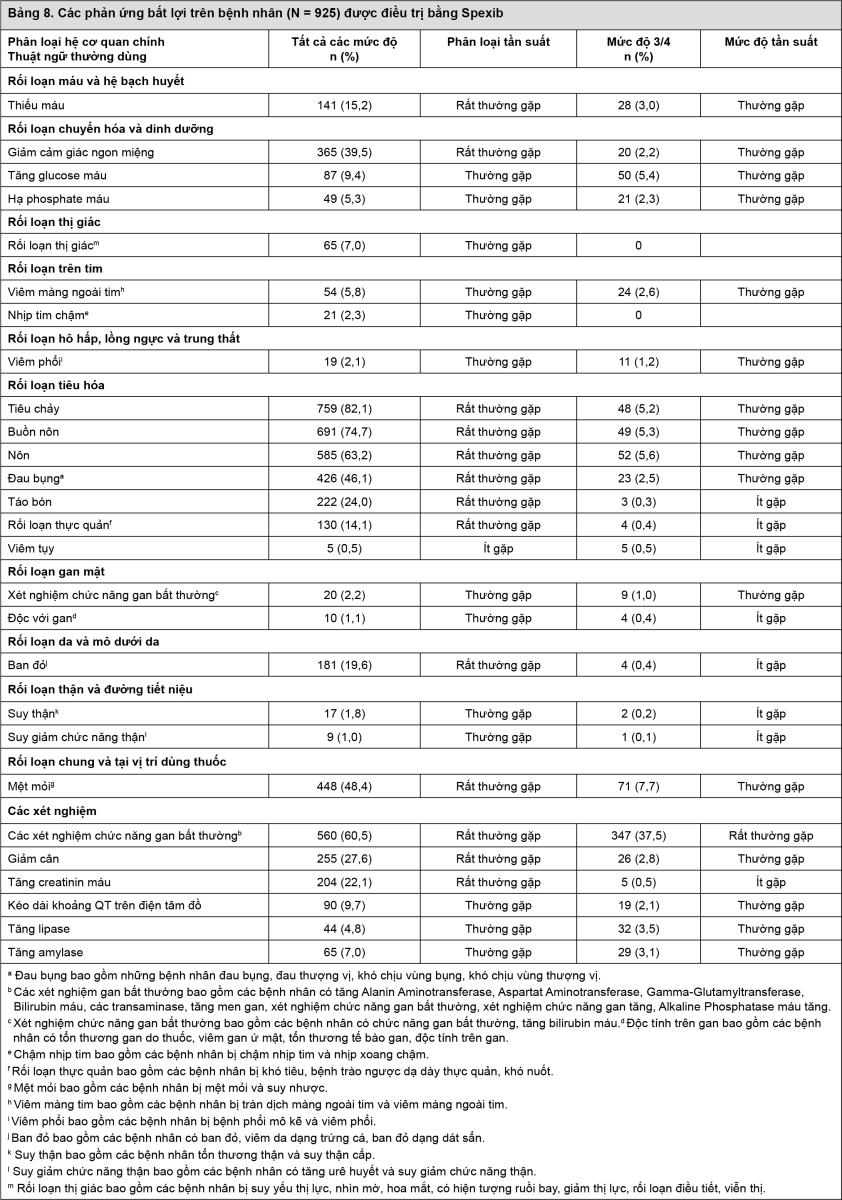
Bảng 8 trình bày các phản ứng bất lợi sắp xếp theo tần suất của Spexib được ghi nhận trên bệnh nhân điều trị với liều khởi đầu 750 mg (N=925) trong 7 nghiên cứu lâm sàng.
Các biến cố bất lợi được liệt kê theo hệ cơ quan theo phân loại MedDRA. Trong mỗi phân nhóm theo hệ cơ quan, các phản ứng bất lợi được sắp xếp theo tần suất xuất hiện, với biến cố bất lợi thường gặp nhất được liệt kê đầu tiên. Ngoài ra, các mức độ tần suất tương ứng sử dụng quy ước dưới đây (CIOMS III) được áp dụng cho từng phản ứng bất lợi: rất thường gặp (≥1/10); thường gặp (≥1/100 đến <1/10); ít gặp (≥1/1.000 đến <1/100); hiếm gặp (≥1/10.000 đến <1/1.000); rất hiếm gặp (<1/10.000) và chưa biết rõ (không ước tính được từ các dữ liệu hiện có).
- xem Bảng 8.
Phản ứng bất lợi trên đường tiêu hóa
Trong một nghiên cứu lâm sàng của Spexib so sánh liều 450 mg hoặc 600 mg mỗi ngày dùng với thức ăn (N=44, N=46) với liều 750 mg mỗi ngày trong điều kiện nhịn ăn (N=45), phản ứng bất lợi trên đường tiêu hóa (tất cả các mức độ phản ứng bất lợi gồm tiêu chảy, nôn và buồn nôn) thấp hơn ở nhóm dùng liều 450 mg với thức ăn (tương ứng 47,7%; 45,5% và 22,7%) so với nhóm nhịn ăn dùng liều 750 mg (tương ứng 64,4%; 62,2% và 42,2%). Các phản ứng bất lợi độ 1 của tiêu chảy, buồn nôn và nôn xảy ra lần lượt là 43,2%; 29,5% và 18,2% đối với bệnh nhân trong nhóm dùng liều 450 mg với thức ăn và 51,1%; 40,0% và 33,3% ở nhóm bệnh nhân nhịn ăn dùng liều 750 mg. Các phản ứng bất lợi độ 2 của tiêu chảy, buồn nôn và nôn xảy ra lần lượt là 4,5%; 15,9% và 4,5% đối với bệnh nhân trong nhóm dùng liều 450 mg với thức ăn và 13,3%; 15,6% và 2,2% ở nhóm bệnh nhân nhịn ăn dùng liều 750 mg. Không có trường hợp bị tiêu chảy, buồn nôn và nôn mửa ở mức độ 3 hoặc 4 ở nhóm dùng liều 450 mg với thức ăn, trong khi tỷ lệ này là 0%; 6,7% và 6,7% trong nhóm nhịn ăn dùng liều 750 mg. Hồ sơ an toàn tổng thể trong nghiên cứu này phù hợp với nghiên cứu đã được thiết lập cho Spexib (xem phần Liều lượng và Cách dùng và Dược lý).
Nhóm bệnh nhân đặc biệt
Trong 7 thử nghiệm lâm sàng, 168 trong số 925 bệnh nhân (18,2%) điều trị bằng Spexib từ 65 tuổi trở lên. Dữ liệu về độ an toàn của thuốc trên bệnh nhân từ 65 tuổi trở lên tương tự trên bệnh nhân dưới 65 tuổi (xem mục Liều lượng và Cách dùng).
Bảo quản
Không bảo quản trên 30°C.
Phân loại ATC
L01XE28 - ceritinib
Trình bày/Đóng gói
Viên nang: hộp 5 vỉ x 10 viên.

- Abacavir
- Abernil
- Abiiogran
- Acarbose
- ACC
- Acebutolol
- Acenocoumarol
- Acetate Ringer's
- Acetazolamide
- Acetylcystein
- Acetylsalicylic acid
- Aciclovir
- Acid acetylsalicylic
- Acid aminocaproic
- Acid ascorbic
- Acid boric
- Acid chenodeoxycholic
- Acid ethacrynic
- Acid folic
- Acid fusidic
- Acid iopanoic
- Acid ioxaglic
- Acid nalidixic
- Acid pantothenic
- Acid para-aminobenzoic
- Acid salicylic
- Acid tranexamic
- Acid valproic
- Acid zoledronic
- Acitretin
- Aclasta
- Aclon
- Actapulgite
- Actelsar
- Actelsar HCT
- Actemra
- Actilyse
- Acular
- Acupan
- Acuvail
- Acyclovir STADA
- Acyclovir STADA Cream
- Adalat
- Adenosin
- Adenosin Ebewe
- Adipiodon
- Advagraf
- Aerius
- Afinitor
- Agicarvir
- Agifovir-E
- Agilosart
- Agilosart-H
- Agimepzol
- Agimosarid
- Agimstan
- Agimstan-H
- Agiremid
- Agivastar
- Aibezym
- Air-X
- Alaxan
- Albendazol
- Albiomin
- Albumin
- Albumin người Grifols 20%
- Albuminar
- AlbuRx
- Albutein
- Alcuronium chloride
- Aldesleukin
- Alendronat
- Alertin
- Alfa-Lipogamma 600 Oral
- Alfuzosin hydrochlorid
- Algotra
- Alimemazin
- Alimta
- Allipem
- Allopurinol
- Allopurinol STADA
- Aloxi
- Alprazolam
- Alpha Chymotrypsin
- Alpha tocopherol
- Alphachymotrypsin Glomed
- Alphagan-P
- Aluvia
- Alzental
- Amaryl
- Ambroco
- Ambroxol
- Amcinol-Paste
- Amigold
- Amikacin
- Aminocaproic acid
- Aminoleban
- Aminoleban Oral
- Aminosteril N-Hepa
- Amiparen
- Amitriptyline
- Amiyu
- Amlodipine
- Amlor
- Amoxicillin
- Amoxicillin & clavulanate
- Ampicillin
- Amquitaz
- Anaferon for children
- Anargil
- Anaropin
- Andriol Testocaps
- Anepzil
- Anyfen
- Apaisac
- Apidra SoloStar
- Apitim 5
- Aprovel
- Aquaphil
- Arcalion
- Arcoxia
- Aricept Evess
- Arimidex
- Arnetine
- Artrodar
- A-Scabs
- Ascorbic acid
- Asperlican/Candinazol
- Aspilets EC
- Aspirin
- Asthmatin
- Atelec
- Atocib 120
- Atocib 90
- Atosiban PharmIdea
- Atozet
- Attapulgite
- Atussin
- Atropin
- Augbactam
- Augmentin Sachet
- Augmentin SR
- Augmentin Tablets
- Augmex
- Avamys
- Avastin
- Avelox Dịch truyền
- Avelox Viên nén
- Avodart
- Axcel Cefaclor-125 Suspension
- Axcel Cetirizine Syrup
- Axcel Chlorpheniramine
- Axcel Dexchlorpheniramine
- Axcel Dicyclomine-S Syrup
- Axcel Diphenhydramine Paediatric Syrup
- Axcel Erythromycin ES
- Axcel Eviline
- Axcel Fungicort Cream
- Axcel Fusidic acid Cream
- Axcel Fusidic acid-B Cream
- Axcel Hydrocortisone Cream
- Axcel Lignocaine 2% Sterile Gel
- Axcel Loratadine
- Axcel Miconazole Cream
- Axcel Paracetamol
- Axcel Urea Cream
- Axitan
- Azenmarol
- Azicine
- Aziphar
- Azithromycin
Quảng cáo




