Nhà sản xuất
Novartis Pharma
Nhà phân phối
Phytopharma
Thành phần
Mỗi viên: Imatinib (dạng mesylate) 100mg.
Mô tả
Viên nén bao phim Glivec 100mg có thể bẻ được: Viên nén bao phim màu vàng rất đậm đến màu cam hơi nâu, hình tròn, có khắc chữ “NVR” và chữ “SA” trên một mặt và một vạch trên mặt kia.
Dược lý
Nhóm dược lý: Thuốc ức chế Protein-tyrosine kinase.
Mã ATC: L01XE01.
Cơ chế tác động: Imatinib là một chất ức chế protein tyrosine kinase phân tử nhỏ, ức chế hoạt động của các BCR-ABL tyrosine kinase (TK) cũng như các thụ thể TK: c-Kit, thụ thể của yếu tố tế bào mầm (SCF) được mã hóa cho các c-Kit proto-oncogene, các thụ thể discoidin miền (DDR1) và (DDR2), thụ thể yếu tố kích thích tạo khúm (CSF-1R) và thụ thể alpha và beta của yếu tố tăng trưởng có nguồn gốc tiểu cầu (PDGFR-alpha và PDGFR-beta). Imatinib cũng có thể ức chế các hoạt động của tế bào qua trung gian hoạt hóa của các thụ thể kinase này.
Dược lực học
Imatinib là một chất ức chế protein tyrosine kinase ức chế mạnh khu vực nhóm điểm gãy-Abelson (BCR-ABL) tyrosine kinase in vitro, trong tế bào, in vivo. Hợp chất này ức chế chọn lọc sự tăng sinh và thúc đẩy quá trình chết tế bào theo chương trình (apoptosis) trong dòng tế bào có BCR-ABL dương tính cũng như những tế bào bệnh bạch cầu mới của những bệnh nhân bị CML với nhiễm sắc thể Philadelphia dương tính và bệnh nhân bị bệnh bạch cầu nguyên bào lymphô cấp (ALL). Trong các nghiệm pháp chuyển dạng khúm, sử dụng các mẫu máu ngoại biên và tủy xương ex vivo, imatinib cho thấy có ức chế chọn lọc các dòng tế bào BCR-ABL dương tính ở những bệnh nhân bị bệnh bạch cầu tủy mạn.
In vivo hợp chất này cho thấy tác dụng chống u như một đơn chất trong các mô hình động vật sử dụng tế bào u có BCR-ABL dương tính.
Imatinib còn là một chất ức chế thụ thể tyrosin kinase đối với yếu tố tăng trưởng có nguồn gốc tiểu cầu (PDGF) và yếu tố tế bào mầm (SCF), c-Kit, và ức chế các phản ứng tế bào qua trung gian PDGF và SCF. In vitro, imatinib ức chế sự tăng sinh và thúc đẩy quá trình chết tế bào theo chương trình trong tế bào u mô đệm dạ dày ruột (GIST), biểu thị một sự đột biến c-Kit hoạt động. Sự hoạt hóa cơ bản PDGFR hoặc ABL protein tyrosin kinase như là hậu quả của sự kết hợp thành các protein khác nhau hoặc sự sản xuất các thành phần của PDGF cho thấy sự liên quan đến sinh bệnh học của MDS/MPD, HES/CEL và DFSP. Hơn nữa sự hoạt hóa cơ bản của c-Kit hoặc PDRFG cho thấy sự liên quan về sinh bệnh học của u tế bào dưỡng bào hệ thống (SM). Imatinib ức chế truyền tín hiệu và tăng sinh tế bào do rối loạn điều chỉnh hoạt động của PDGFR, c-Kit và ABL kinase.
CÁC NGHIÊN CỨU LÂM SÀNG
Những nghiên cứu lâm sàng trong bệnh CML
Hiệu quả của Glivec dựa trên tỷ lệ đáp ứng nói chung về huyết học và di truyền học tế bào và thời gian còn sống không có tiến triển bệnh.
Ba nghiên cứu giai đoạn II lớn, đa quốc gia, nhãn mở, không đối chứng đã được tiến hành trên những bệnh nhân bị bệnh bạch cầu tủy mạn (CML) với nhiễm sắc thể Philadelphia dương tính (Ph+) giai đoạn tiến triển, cơn nguyên bào hoặc giai đoạn tăng tốc, hoặc bệnh bạch cầu có Ph+ khác hoặc bệnh bạch cầu tủy mạn giai đoạn mạn tính nhưng thất bại với điều trị bằng interfon-alpha (IFN). Một nghiên cứu lớn ngẫu nhiên, giai đoạn III nhãn mở, đa trung tâm, đa quốc gia đã được tiến hành trên những bệnh nhân bị Ph+ CML mới được chẩn đoán. Ngoài ra, trẻ em đã được điều trị trong 2 nghiên cứu giai đoạn I và một thử nghiệm giai đoạn II đa trung tâm, nhãn mở, đơn nhóm.
Trong tất cả nghiên cứu lâm sàng, có 38-40% bệnh nhân ≥60 tuổi và 10-12% bệnh nhân ≥ 70 tuổi.
Giai đoạn mạn tính, mới được chẩn đoán
Nghiên cứu giai đoạn III này so sánh việc điều trị đơn thuần bằng Glivec với liều 400 mg/ngày hoặc interferon-alpha (IFN) 5 MIU/m2/ngày phối hợp với cytarabine (Ara-C) 20 mg/m2/ngày, cùng tiêm dưới da 10 ngày trong một tháng. Các bệnh nhân có biểu hiện thiếu đáp ứng (thiếu đáp ứng huyết học hoàn toàn (CHR) sau 6 tháng, tăng bạch cầu (WBC), không có đáp ứng tốt về di truyền học tế bào (MCyR) sau 24 tháng), mất đáp ứng (mất CHR hoặc McyR) hoặc dung nạp rất kém với điều trị được phép đổi sang nhóm điều trị khác.
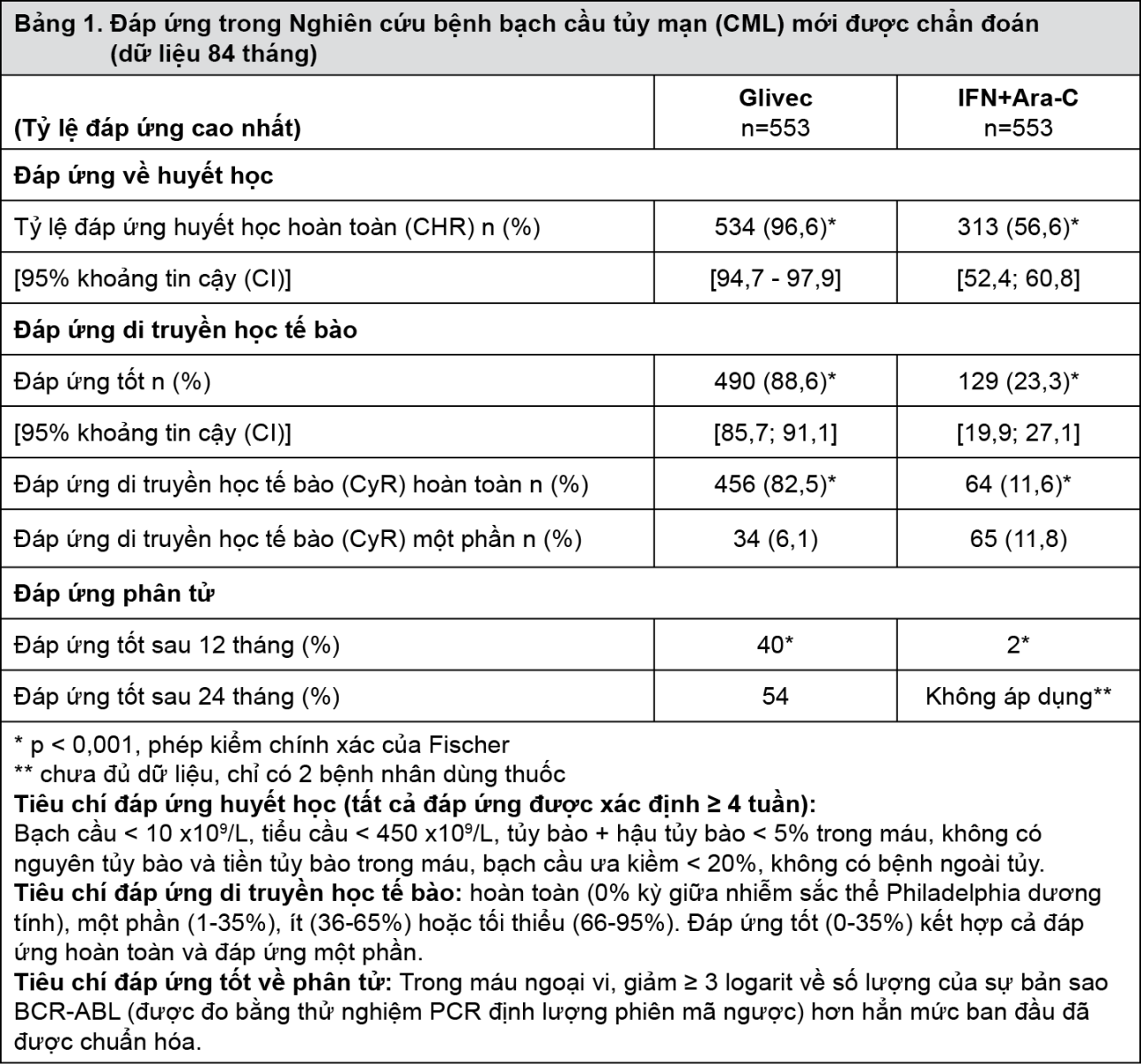
Tổng số 1.106 bệnh nhân đã được chọn ngẫu nhiên, 533 bệnh nhân cho mỗi nhóm. Tuổi trung vị là 51 tuổi (từ 18 đến 70 tuổi), với 21,9% bệnh nhân ≥60 tuổi. Có 59% nam và 41% nữ. Sau 7 năm theo dõi, thời gian trung vị của trị liệu bước một là 82 tháng ở nhóm Glivec và 8 tháng ở nhóm IFN. Thời gian trung vị của trị liệu bước hai bằng Glivec là 64 tháng. Nói chung, ở những bệnh nhân dùng Glivec bước một, liều trung bình hàng ngày của Glivec là 406±78 mg. Do tỷ lệ ngừng điều trị và chuyển điều trị đều cao hơn, chỉ 2% bệnh nhân được phân ngẫu nhiên dùng IFN tiếp tục dùng thuốc này bước một. Trong nhóm dùng IFN, rút lại cam kết nghiên cứu (14%) là lý do thường gặp nhất đối với việc ngừng trị liệu bước một, và lý do thường gặp nhất đối với việc chuyển sang nhóm Glivec là dung nạp rất kém với điều trị (26%) và do tiến triển của bệnh (14%). Kết cục chính về hiệu quả của nghiên cứu này là tỷ lệ bệnh nhân còn sống còn không tiến triển bệnh. Sự tiến triển được xác định khi có bất kỳ sự cố nào sau đây: tiến triển đến giai đoạn tăng tốc hoặc cơn nguyên bào (AP/BC), tử vong, mất đáp ứng huyết học hoàn toàn (CHR) hoặc không có đáp ứng tốt về di truyền tế bào học (MCyR), hoặc ở bệnh nhân không đạt được đáp ứng huyết học hoàn toàn mà có tăng bạch cầu mặc dù đã có biện pháp điều trị thích hợp. Đáp ứng tốt về di truyền học tế bào, đáp ứng huyết học, đáp ứng phân tử (đánh giá bệnh còn lại ở mức tối thiểu), thời gian đến giai đoạn tăng tốc hoặc cơn nguyên bào và tỷ lệ bệnh nhân còn sống là những kết cục phụ chủ yếu. Dữ liệu về đáp ứng được biểu thị trong bảng 1.
- xem Bảng 1.
Sau 7 năm theo dõi, có 93 trường hợp có biến cố tiến triển bệnh (16,8%) ở nhóm dùng Glivec: 37 trường hợp (6,7%) tiến triển đến giai đoạn tăng tốc hoặc cơn nguyên bào (AP/BC), 31 trường hợp (5,6%) mất đáp ứng tốt di truyền học tế bào (MCyR), 15 trường hợp (2,7%) mất đáp ứng huyết học hoàn toàn hoặc tăng bạch cầu và 10 trường hợp (1,8%) tử vong không liên quan đến bệnh bạch cầu mạn. Ngược lại, có 165 trường hợp (29,8%) trong nhóm dùng interferon + cytarabin (IFN + Ara-C) trong đó 130 trường hợp xảy ra trong suốt thời gian trị liệu bước một bằng IFN + Ara-C.
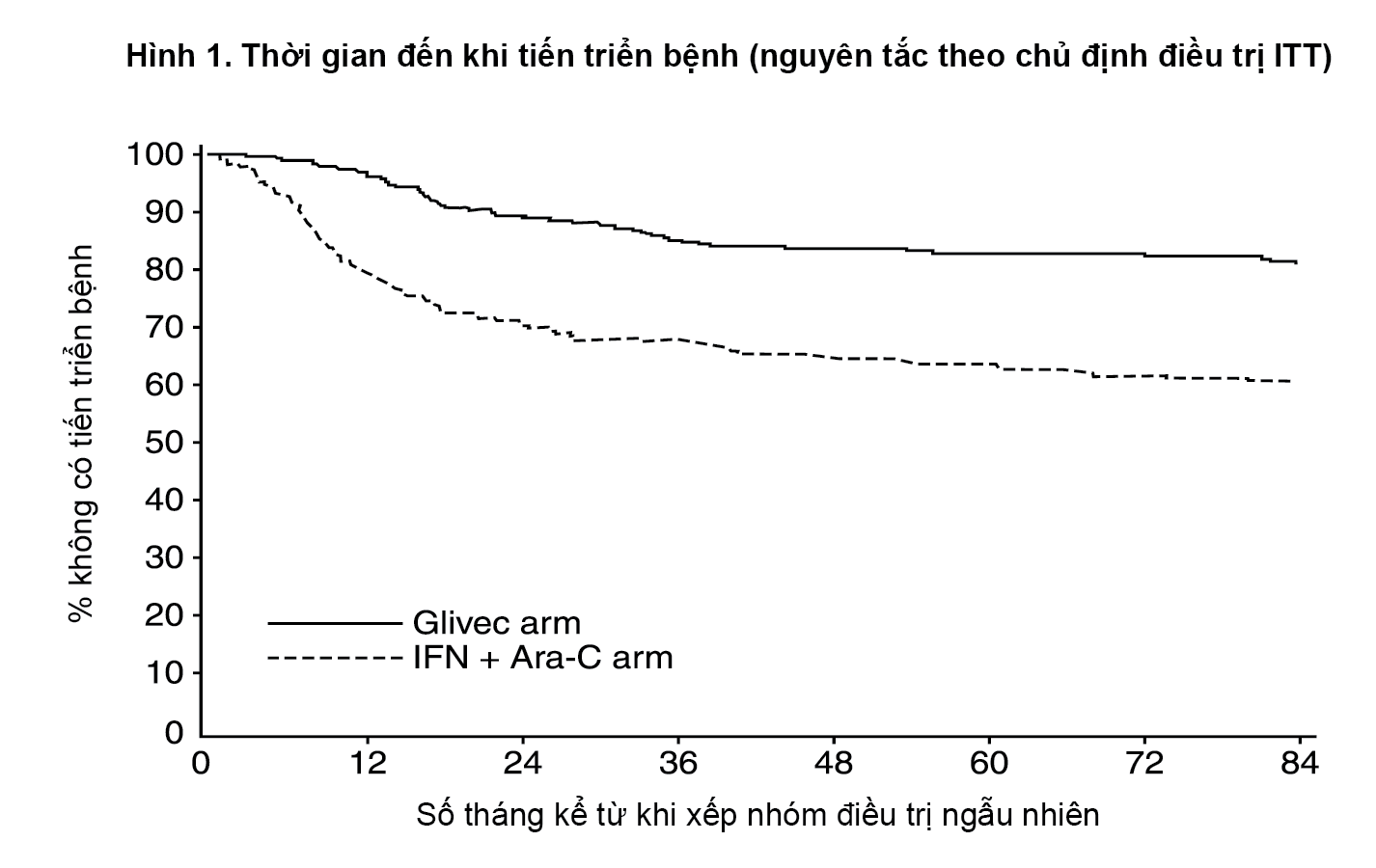
Tỷ lệ ước tính về số bệnh nhân còn sống không tiến triển bệnh sau 84 tháng là 81,2% với khoảng tin cậy 95% (78-85) ở nhóm dùng Glivec và 60,6% (56-65) ở nhóm chứng (P<0,001) (Hình 1). Tỷ lệ tiến triển hàng năm đối với Glivec là 3,3% trong năm đầu sau khi bắt đầu nghiên cứu, 7,5% trong năm thứ hai, 4,8% trong năm thứ ba, 1,7% trong năm thứ tư, 0,8% trong năm thứ năm, 0,3% trong năm thứ sáu và 2% trong năm thứ bảy của cuộc nghiên cứu.
Tỷ lệ ước tính bệnh nhân không có tiến triển đến giai đoạn tăng tốc hoặc cơn nguyên bào sau 84 tháng cao hơn đáng kể ở nhóm dùng Glivec so với nhóm dùng interferon (92,5% so với 85,1%, p<0,001).
- xem Hình 1.
Tổng số có 71 bệnh nhân tử vong (12,8%) ở nhóm dùng Glivec và 85 bệnh nhân tử vong (15,4%) ở nhóm dùng interferon + cytarabine (IFN+Ara-C). Sau 84 tháng tỷ lệ sống còn toàn bộ ước tính là 86,4% (KTC 95% 83-90) ở nhóm chọn ngẫu nhiên dùng Glivec so với 83,3% (KTC 95% 80-87) ở nhóm dùng IFN+Ara-C (p=0,073, phép kiểm log-rank). Kết cục về thời gian đến khi có biến cố này chịu ảnh hưởng mạnh bởi tỷ lệ cao bệnh nhân chuyển nhóm điều trị từ IFN+Ara-C sang Glivec. Khi cắt số liệu 48 trường hợp tử vong xảy ra sau khi ghép tủy xương, tỷ lệ bệnh nhân còn sống sau 84 tháng là 89,6% so với 88,1% (p=0,200, phép kiểm log-rank). Chỉ có 31 trường hợp tử vong (5,6%) (trước khi ghép tủy xương) ở bệnh nhân dùng Glivec được qui cho bệnh CML, so với 40 bệnh nhân tử vong (7,2%) dùng IFN+Ara-C. Nếu chỉ xem xét số bệnh nhân tử vong liên quan đến bệnh bạch cầu tủy mạn và xem xét mọi trường hợp tử vong sau khi ghép tủy xương hoặc do nguyên nhân nào khác, tỷ lệ ước tính bệnh nhân còn sống sau 84 tháng là 93,6% so với 91,1% (p=0,1, phép kiểm log-rank).
Trong nghiên cứu này, cho phép tăng liều từ 400 mg/ngày lên 600 mg/ngày sau đó từ 600 mg/ngày đến 800 mg/ngày. Sau 42 tháng theo dõi, 11 bệnh nhân đã đạt được đáp ứng huyết học hoàn toàn (CHR) sau 3 tháng, và đáp ứng tốt di truyền học tế bào (MCyR) sau 12 tháng trong khi dùng liều 400 mg/ngày đã được xác định là bị mất đáp ứng di truyền học tế bào (trong vòng 4 tuần). Trong số 11 bệnh nhân này, 4 bệnh nhân tăng đến 800 mg/ngày, 2 trong số này đạt lại đáp ứng di truyền học tế bào (1 bệnh nhân đáp ứng một phần và 1 bệnh nhân đáp ứng hoàn toàn, trường hợp sau còn đạt được đáp ứng phân tử), trong khi trong số 7 bệnh nhân không tăng liều, chỉ có 1 bệnh nhân đạt lại đáp ứng di truyền học tế bào hoàn toàn. Tỷ lệ phần trăm của một số phản ứng phụ (ADRs) cao hơn ở 40 bệnh nhân với liều tăng đến 800 mg/ngày so với nhóm bệnh nhân trước khi tăng liều (n=551). Những phản ứng phụ thường gặp bao gồm xuất huyết tiêu hóa, viêm kết mạc và tăng transaminase hoặc bilirubin. Các phản ứng phụ khác được báo cáo với tần suất thấp hơn hoặc tương đương.
Giai đoạn mạn tính, thất bại với điều trị interferon
532 bệnh nhân được điều trị với liều khởi đầu 400 mg. Bệnh nhân được phân ra làm 3 loại chính: thất bại về huyết học (29%), thất bại về di truyền học tế bào (35%) hoặc không dung nạp với interferon (36%). Các bệnh nhân đã được điều trị trung bình 14 tháng trước đó bằng interferon với liều ≥ 25x106 IU/tuần và tất cả đều ở cuối giai đoạn mạn tính, với trung vị thời gian kể từ lúc chẩn đoán là 32 tháng. Biến số hiệu quả chính của nghiên cứu là tỷ lệ của đáp ứng tốt về di truyền học tế bào (đáp ứng hoàn toàn cộng với đáp ứng một phần, 0-35% kỳ giữa của phân bào với nhiễm sắc thể Philadelphia dương tính ở tủy xương).
Trong nghiên cứu này, 65% bệnh nhân đạt được đáp ứng tốt về di truyền học tế bào (MCyR) trong đó 53% bệnh nhân đáp ứng hoàn toàn. Đáp ứng huyết học hoàn toàn (CHR) đạt được ở 95% bệnh nhân.
Giai đoạn tăng tốc
235 bệnh nhân ở giai đoạn cấp của bệnh được tham gia nghiên cứu. Đầu tiên 77 bệnh nhân được bắt đầu với 400 mg và 158 bệnh nhân còn lại được bắt đầu với liều 600 mg.
Biến số hiệu quả chính là tỷ lệ đáp ứng huyết học, được ghi nhận hoặc là đáp ứng huyết học hoàn toàn (CHR), hoặc không còn dấu hiệu của bệnh bạch cầu (tức là không còn nguyên bào ở tủy xương và máu nhưng không có sự phục hồi máu ngoại vi đầy đủ như đối với đáp ứng hoàn toàn), hoặc là trở lại bệnh bạch cầu tủy mạn giai đoạn mạn tính. Đáp ứng huyết học xác định đã đạt được ở 71,5% bệnh nhân. Điều quan trọng là 27,7% bệnh nhân cũng đạt được đáp ứng tốt về di truyền học tế bào (MCyR) trong đó đáp ứng hoàn toàn chiếm 20,4% số bệnh nhân. Đối với những bệnh nhân được điều trị 600 mg, ước tính trung vị thời gian còn sống không tiến triển là 22,9 tháng và thời gian sống còn toàn bộ là 42,5 tháng. Trong một phân tích đa biến số, liều 600 mg có liên quan với cải thiện thời gian tiến triển bệnh, không phụ thuộc vào số lượng tiểu cầu, nguyên bào máu và hemoglobin ≥10 g/L.
Giai đoạn chuyển cấp cơn nguyên bào tủy
260 bệnh nhân có cơn nguyên bào tủy được tham gia nghiên cứu. 95 bệnh nhân (37%) đã được dùng hóa trị liệu trước đó để điều trị giai đoạn tăng tốc hoặc cơn nguyên bào (“những bệnh nhân đã được điều trị trước”) trong khi 165 bệnh nhân (63%) chưa được điều trị (“những bệnh nhân chưa được điều trị"). Ba mươi bảy bệnh nhân đầu tiên được bắt đầu với liều 400 mg và 223 bệnh nhân còn lại được bắt đầu với liều 600 mg.
Biến số hiệu quả chính là tỷ lệ của đáp ứng huyết học, được báo cáo hoặc là đáp ứng huyết học hoàn toàn (CHR), hoặc không còn dấu hiệu của bệnh bạch cầu, hoặc là trở lại bệnh bạch cầu tủy mạn giai đoạn mạn tính, 31% bệnh nhân đạt được đáp ứng huyết học (36% ở nhóm bệnh nhân chưa được điều trị và 22% ở nhóm bệnh nhân đã được điều trị trước). Tỷ lệ đáp ứng cũng cao hơn ở những bệnh nhân được điều trị liều 600 mg (33%) khi so sánh với những bệnh nhân được điều trị liều 400 mg (16%, p=0,022). Ước tính trung vị thời gian sống còn của những bệnh nhân chưa được điều trị là 7,7 tháng và bệnh nhân đã được điều trị trước là 4,7 tháng.
Bệnh nhi
Tổng số 51 bệnh nhi bị bệnh bạch cầu tủy mạn ở giai đoạn mạn tính mới được chẩn đoán và chưa được điều trị đã được tham gia nghiên cứu trong một thử nghiệm giai đoạn II đơn nhóm, nhãn mở, đa trung tâm và được điều trị bằng Glivec 340 mg/m2/ngày. Điều trị Glivec đã tạo ra đáp ứng nhanh chóng ở bệnh nhi bị bệnh bạch cầu tủy mạn mới được chẩn đoán với đáp ứng huyết học hoàn toàn (CHR) là 78% sau 8 tuần điều trị và tỷ lệ về đáp ứng di truyền học tế bào hoàn toàn (CCyR) là 65% (tương đương với kết quả quan sát được ở người lớn) sau 3 đến 10 tháng điều trị.
Tổng số 31 bệnh nhi đã được điều trị tích cực trước đó (45% bệnh nhi đã được điều trị trước đó bằng ghép tủy xương (BMT) và 68% được dùng đa hóa trị liệu) bị bệnh bạch cầu tủy mạn giai đoạn mạn tính (n=15) hoặc bệnh bạch cầu tủy mạn trong cơn nguyên bào hoặc bệnh Ph+ALL (n=16) được tham gia nghiên cứu trong thử nghiệm giai đoạn I có tăng liều. Các bệnh nhân được điều trị Glivec với liều dao động từ 260 mg/m2/ngày đến 570 mg/m2/ngày. Trong số 13 bệnh nhân bị bệnh bạch cầu tủy mạn và có các dữ liệu về di truyền học tế bào, 7 bệnh nhân (54%) đạt được đáp ứng di truyền học tế bào hoàn toàn và 4 bệnh nhân (31%) đạt được đáp ứng di truyền học tế bào một phần đối với tỷ lệ đáp ứng tốt về di truyền học tế bào (MCyR) là 85%.
Các nghiên cứu lâm sàng trong bệnh Ph+ ALL: Tổng số 851 bệnh nhân bị bệnh bạch cầu lymphô cấp với nhiễm sắc thể Philadelphia dương tính mới được chẩn đoán hoặc bệnh tái phát/đề kháng tham gia trong 11 nghiên cứu lâm sàng, trong đó 10 nghiên cứu không đối chứng và 1 nghiên cứu ngẫu nghiên. Trong số 851 bệnh nhân, có 93 bệnh nhân trẻ em (bao gồm 4 bệnh nhân trên 18 tuổi và dưới 22 tuổi) đã được điều trị trong một nghiên cứu pha III, không chia ngẫu nhiên, đa trung tâm, nhãn mở.
Bệnh Ph+ ALL mới được chẩn đoán
Trong một nghiên cứu có đối chứng (ADE10) dùng Glivec so với hóa trị tấn công ở 55 bệnh nhân mới được chẩn đoán có tuổi từ 55 tuổi trở lên, Glivec được dùng như một thuốc duy nhất tạo ra tỷ lệ đáp ứng huyết học hoàn toàn cao hơn đáng kể so với hóa trị liệu (96,3% so với 50%; p=0,0001). Khi điều trị cứu vớt bằng Glivec cho những bệnh nhân không đáp ứng hoặc đáp ứng kém với hóa trị liệu, đã có đến 9 bệnh nhân (81,8%) trong số 11 bệnh nhân đạt được đáp ứng huyết học hoàn toàn. Hiệu quả lâm sàng này có kết hợp với sự giảm phiên mã BCR-ABL nhiều hơn ở các bệnh nhân được điều trị bằng Glivec so với nhóm dùng hóa trị liệu sau 2 tuần điều trị (p=0,02). Tất cả bệnh nhân dùng Glivec và củng cố bằng hóa trị liệu sau khi điều trị tấn công và mức độ phiên mã BCR-ABL giống nhau ở cả 2 nhóm sau 8 tuần. Như dự kiến trên cơ sở thiết kế nghiên cứu, không quan sát thấy sự khác nhau nào về thời gian lui bệnh, thời gian sống còn không bệnh hoặc sống còn toàn bộ, mặc dù bệnh nhân có đáp ứng phân tử hoàn toàn và bệnh nhân còn bệnh tối thiểu đã có kết cục tốt hơn về cả thời gian lui bệnh (p=0,01) và thời gian sống còn không bệnh (p=0,02).
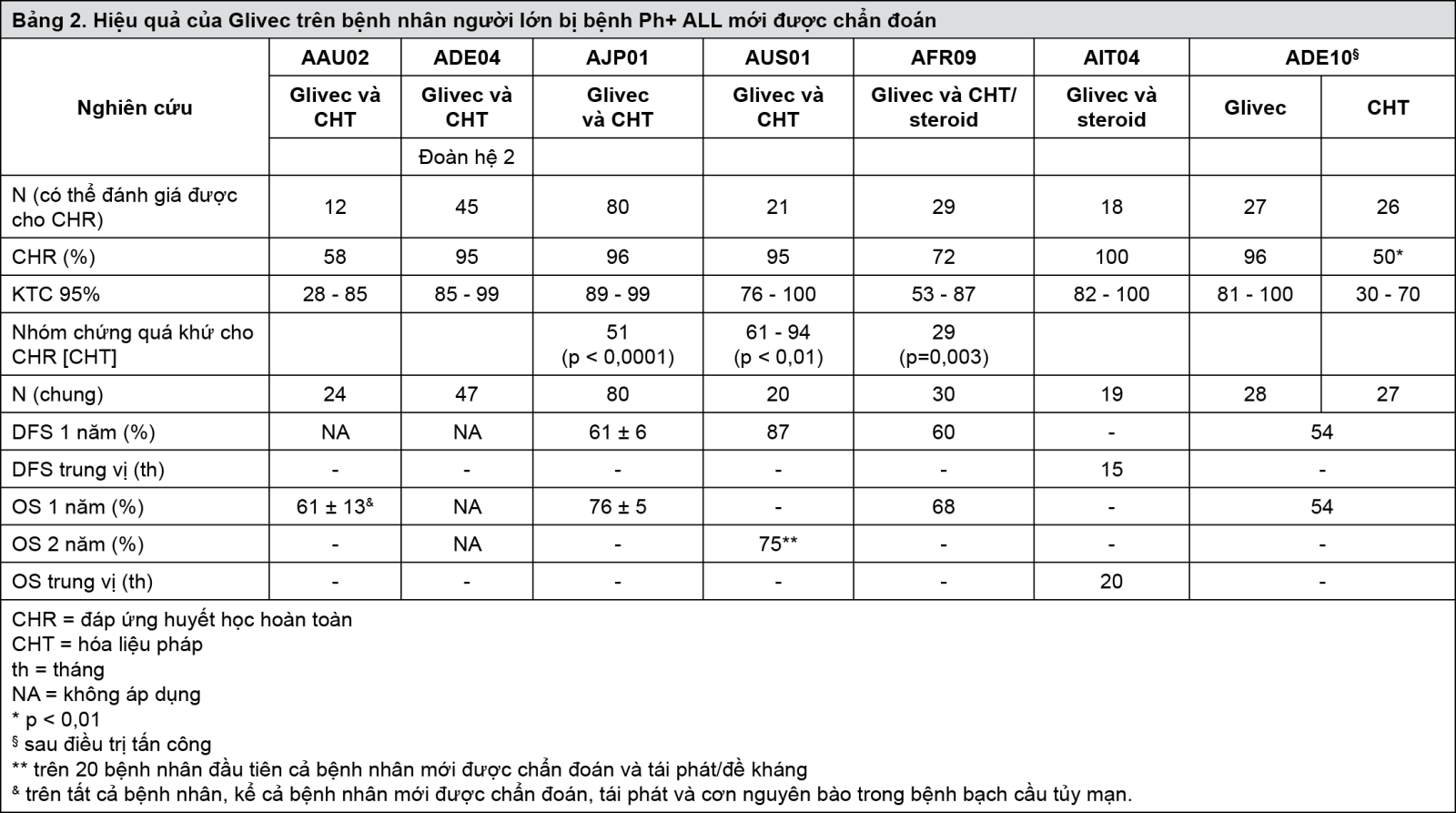
Các kết quả quan sát được trong nhóm 211 bệnh nhân bị bệnh bạch cầu nguyên bào lymphô cấp với nhiễm sắc thể Philadelphia dương tính mới được chẩn đoán trong 4 nghiên cứu lâm sàng không đối chứng (AAU02, ADE04, AJP01 và AUS01) phù hợp với các kết quả đã mô tả trên. Tương tự, trong 2 nghiên cứu lâm sàng không đối chứng (AFR09 và AIT04), 49 bệnh nhân từ 55 tuổi trở lên bị bệnh bạch cầu nguyên bào lymphô cấp với nhiễm sắc thể Philadelphia dương tính (Ph+ ALL) mới được chẩn đoán được cho dùng Glivec kết hợp với steroid có hoặc không dùng hóa trị liệu. Kết quả được thể hiện ở bảng 2.
- xem Bảng 2.
Bệnh nhi: Trong nghiên cứu I2301, có tổng số 93 bệnh nhi, thiếu niên và bệnh nhân trẻ tuổi (bao gồm 4 bệnh nhân trên 18 tuổi và dưới 22 tuổi) bị bệnh Ph+ALL được thu nhận vào một thử nghiệm lâm sàng pha III không ngẫu nhiên, thuần tập tuần tự nhãn mở, đa trung tâm, và được điều trị với Glivec (340 mg/m2/ngày) kết hợp với hóa trị liệu tăng cường sau điều trị tấn công. Glivec được dùng gián đoạn ở các đoàn hệ 1 đến 5, với việc khởi đầu sớm hơn và tăng thời gian dùng Glivec từ đoàn hệ này đến đoàn hệ khác; đoàn hệ 1 nhận cường độ điều trị với Glivec thấp nhất và đoàn hệ 5 nhận cường độ cao nhất (thời gian dài nhất theo ngày với liều Glivec hàng ngày dùng liên tục trong liệu trình hóa trị liệu đầu tiên). Sử dụng Glivec hàng ngày liên tục sớm trong quá trình điều trị kết hợp với hóa trị liệu ở các bệnh nhân đoàn hệ 5 (n=50) cải thiện tỉ lệ sống không biến cố 4 năm (EFS) so với nhóm chứng (n=120), những bệnh nhân nhận hóa trị liệu tiêu chuẩn, không dùng Glivec (tương ứng là 69,6% so với 31,6%). OS 4 năm ước tính ở các bệnh nhân đoàn hệ 5 là 83,6% so với 44,8% ở nhóm chứng trong quá khứ.
Bệnh Ph+ ALL tái phát/đề kháng
Khi Glivec được dùng đơn trị liệu trên bệnh nhân bị bệnh Ph+ ALL tái phát/đề kháng, có 66 bệnh nhân trong số 429 bệnh nhân có thể đánh giá được về đáp ứng, có tỷ lệ đáp ứng huyết học là 33% (12% hoàn toàn) và tỷ lệ đáp ứng tốt di truyền học tế bào là 23%. Trung vị thời gian đến tiến triển trong toàn bộ dân số nghiên cứu gồm 429 bệnh nhân bị bệnh bạch cầu nguyên bào lymphô cấp với nhiễm sắc thể Philadelphia dương tính tái phát/đề kháng nằm trong khoảng từ 1,9 đến 3,1 tháng, và trung vị thời gian sống còn toàn bộ trên 409 bệnh nhân có thể đánh giá được khoảng từ 5 đến 9 tháng. Trong 14 bệnh nhân, dùng Glivec kết hợp với hóa trị liệu tấn công có tỷ lệ đáp ứng huyết học hoàn toàn là 92% ở 12 bệnh nhân có thể đánh giá được và tỷ lệ đáp ứng tốt di truyền học tế bào là 100% ở 8 bệnh nhân có thể đánh giá được. Đáp ứng phân tử đã được đánh giá trên 4 bệnh nhân và 2 bệnh nhân đã đáp ứng hoàn toàn.
Tổng số 14 trong số 146 bệnh nhân đã được điều trị bằng Glivec 600 mg/ngày và có thể đánh giá được về đáp ứng; ghi nhận đáp ứng huyết học hoàn toàn ở 5 bệnh nhân (35%) và đáp ứng tốt di truyền học tế bào ở 7 bệnh nhân (50%). Đáng lưu ý là 4 bệnh nhân được điều trị với liều Glivec thấp hơn (400 mg/ngày) đã không đáp ứng. Trong toàn bộ dân số nghiên cứu gồm 146 bệnh nhân, trung vị thời gian còn sống không bệnh trong khoảng từ 2,8 đến 3,1 tháng và trung vị thời gian sống còn toàn bộ từ 7,4 đến 8,9 tháng.
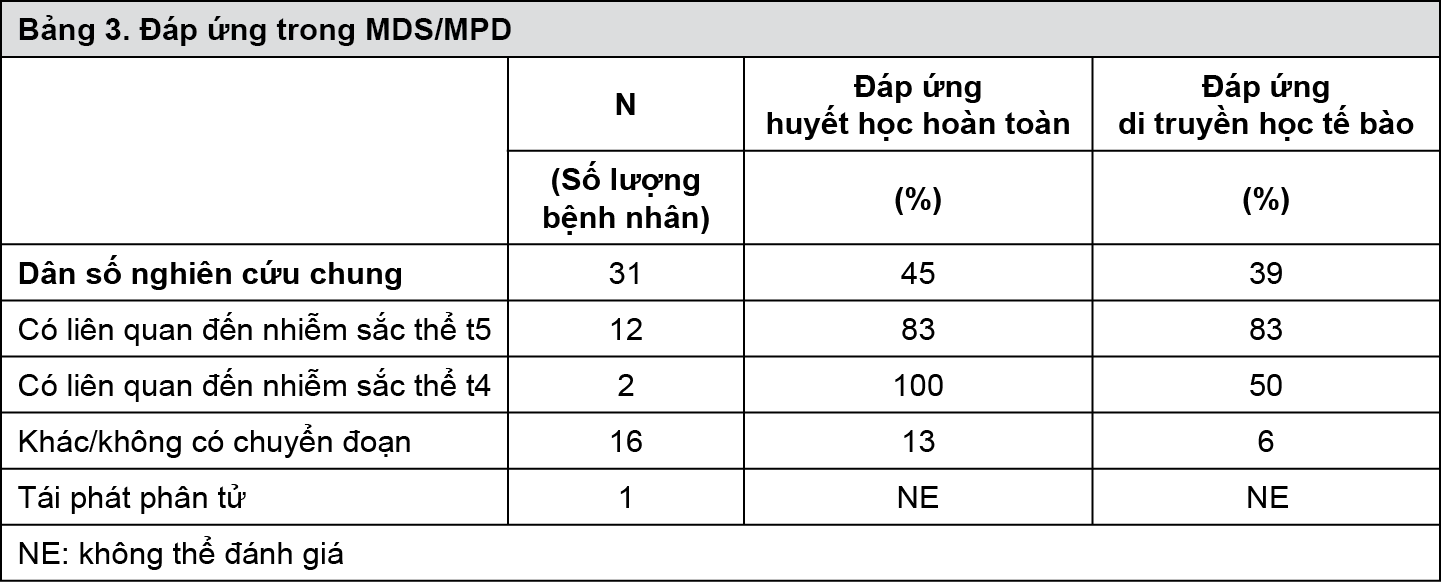
Nghiên cứu lâm sàng trong bệnh MDS/MPD: Một nghiên cứu lâm sàng giai đoạn II, đa trung tâm, nhãn mở (nghiên cứu B2225) tiến hành thử nghiệm Glivec trên những nhóm bệnh nhân khác nhau bị những bệnh đe dọa đến tính mạng có liên quan tới ABL, c-Kit hoặc PDGFR protein tyrosine kinase. Nghiên cứu này bao gồm 7 bệnh nhân bị MDS/MPD trong tổng số 185 bệnh nhân được điều trị, 45 trong số họ có bệnh về máu và 140 khối u đặc các loại. Những bệnh nhân này được điều trị với Glivec 400 mg/ngày. Tuổi của các bệnh nhân trong khoảng từ 20-86. Thêm 24 bệnh nhân bị MDS/MPD tuổi từ 2-79 được tường trình trong 12 báo cáo đã được công bố và 1 thử nghiệm lâm sàng. Những bệnh nhân này cũng được dùng Glivec với liều 400 mg/ngày với 3 bệnh nhân ngoại lệ dùng liều thấp hơn. Trong tổng số 31 bệnh nhân được điều trị bệnh MDS/MPD, 14 (45%) đạt được đáp ứng huyết học hoàn toàn và 9 (29%) đạt được đáp ứng di truyền học tế bào hoàn toàn (39% bao gồm cả các đáp ứng tốt và một phần). Đáng lưu ý là bệnh lý ác tính gây chuyển đoạn thường ảnh hưởng đến nhiễm sắc thể t5q33 hoặc t4q12 dẫn đến sự tái sắp xếp gen PDGFR ở 14 bệnh nhân có thể đánh giá được. Tất cả những bệnh nhân này đã đáp ứng về mặt huyết học (12 có đáp ứng hoàn toàn). Đáp ứng di truyền học tế bào được đánh giá trên 11 trong số 14 bệnh nhân, tất cả trong số họ có đáp ứng (9 bệnh nhân đáp ứng hoàn toàn). Chỉ 2 (13%) trong số 16 bệnh nhân không có chuyển đoạn liên quan đến sự tái sắp xếp gen PDGFR đạt được đáp ứng huyết học hoàn toàn và 1 (6%) đạt được đáp ứng tốt về di truyền học tế bào. Thêm 1 bệnh nhân có sự tái sắp xếp gen PDGFR trong tình trạng tái phát về mặt phân tử sau khi ghép tủy xương đã có đáp ứng về mặt phân tử. Trung vị thời gian điều trị là 12,9 tháng (0,8-26,7) ở 7 bệnh nhân đã được điều trị trong nghiên cứu B2225 và dao động trong khoảng 1 tuần đến hơn 18 tháng ở những bệnh nhân có đáp ứng trong các y văn đã được công bố. Kết quả được đưa ra trong bảng 3.
- xem Bảng 3.
Nghiên cứu lâm sàng trong bệnh SM: Nghiên cứu B2225 cũng bao gồm 5 bệnh nhân (tuổi từ 49 đến 74) bị u dưỡng bào hệ thống (SM) điều trị bằng Glivec 100 mg đến 400 mg/ngày. Có thêm 25 bệnh nhân bị SM tuổi từ 26 đến 85 cũng được dùng Glivec với liều 100 mg đến 400 mg/ngày đã được báo cáo (10 báo cáo ca bệnh và loạt ca bệnh đã được công bố. Trong số 30 bệnh nhân bị SM, 10 (33%) đạt được đáp ứng huyết học hoàn toàn và 9 (30%) đáp ứng huyết học một phần (tỉ lệ đáp ứng chung 63%). Các bất thường về di truyền học tế bào được đánh giá trên 21 trong số 30 bệnh nhân được điều trị trong các báo cáo đã được công bố và trong nghiên cứu B2225. Tám trong số 21 bệnh nhân có FIP1L1-PDGFR-alpha fusion kinase, là loại fusion kinase thường có ở nam giới, bị hoặc không bị tăng bạch cầu ưa eosin. Hai bệnh nhân có đột biến c-Kit ở vùng gần màng (một tại Phe522Cys và một tại K509I). Mười sáu bệnh nhân không biết hoặc không phát hiện được bất thường về di truyền tế bào học. Bốn bệnh nhân có đột biến D816V (một bệnh nhân có đáp ứng mắc kết hợp CML và SM). Phần lớn các bệnh nhân trong y văn có đột biến D816V c-Kit không được xem là nhạy cảm với Glivec. Trung vị thời gian điều trị là 13 tháng (từ 1,4-22,3 tháng) ở 5 bệnh nhân trong nghiên cứu B2225 và trong khoảng từ 1 đến hơn 30 tháng ở bệnh nhân có đáp ứng trong y văn. Kết quả được trình bày trong bảng 4.
- xem Bảng 4.
Nghiên cứu lâm sàng trong HES/CEL: Nghiên cứu B2225 này cũng bao gồm 14 bệnh nhân (tuổi từ 16 đến 64) bị HES/CEL điều trị bằng Glivec 100 mg đến 1000 mg/ngày. Thêm 162 bệnh nhân HES/CEL (tuổi từ 11 đến 78) dùng Glivec với liều 75 mg đến 800 mg/ngày được báo cáo (35 báo cáo ca bệnh và loạt ca bệnh đã được công bố). Trong số 176 bệnh nhân HES/CEL được điều trị, 107 (61%) đạt được đáp ứng huyết học hoàn toàn và 16 (9%) đáp ứng huyết học một phần (tỉ lệ đáp ứng chung 70%). Các bất thường về di truyền học tế bào được đánh giá ở 117 trong số 176 bệnh nhân được điều trị trong các báo cáo đã được công bố và trong nghiên cứu B2225. FIP1L1-PDGFR-alpha fusion kinase được tìm thấy ở 61 trên tổng só 117 bệnh nhân. Tất cả những bệnh nhân dương tính với FIP1L1-PDGFR-alpha fusion kinase này đều đạt được đáp ứng huyết học hoàn toàn. 115 bệnh nhân âm tính với hoặc không biết có FIP1L1-PDGFR-alpha fusion kinase, trong đó 62 (54%) đạt được đáp ứng hoặc là đáp ứng huyết học hoàn toàn (n=46) hoặc một phần (n=16). Kết quả được trình bày trong bảng 5.
- xem Bảng 5.

Ngoài ra, sự cải thiện về triệu chứng học và các bất thường rối loạn chức năng cơ quan khác (tim, thần kinh, da/mô dưới da, hô hấp/ngực/trung thất, cơ xương/mô liên kết/mạch và hệ cơ quan dạ dày ruột) đã được tường trình trong các báo cáo ca bệnh.
Nghiên cứu lâm sàng trên bệnh GIST không thể cắt bỏ hoặc đã di căn
Hai nghiên cứu giai đoạn III, đa quốc gia, ngẫu nhiên, nhãn mở (SWOG, EORTC) đã được tiến hành trên những bệnh nhân bị u mô đệm dạ dày ruột (GIST) ác tính không thể cắt bỏ hoặc đã di căn. Tổng số 1.640 bệnh nhân được chọn ngẫu nhiên 1:1 dùng thuốc liều 400 mg hoặc 800 mg đường uống 1 lần/ngày liên tục cho đến khi bệnh tiến triển hoặc độc tính nặng. Bệnh nhân được phép chuyển sang nhóm điều trị 800 mg 1 lần/ngày. Các nghiên cứu được thiết kế để so sánh tỉ lệ đáp ứng, thời gian sống còn không tiến triển bệnh và thời gian sống còn toàn bộ giữa các nhóm liều. Các bệnh nhân đều có chẩn đoán bệnh học là GIST ác tính có CD 117 dương tính không thể cắt bỏ và/hoặc đã di căn.
Mục tiêu chính của hai nghiên cứu là đánh giá hoặc là thời gian sống còn không tiến triển bệnh (PFS) với mục tiêu phụ là thời gian sống còn toàn bộ (OS) trong một nghiên cứu (EORTC), hoặc là thời gian sống còn toàn bộ với mục tiêu phụ là PFS trong một nghiên cứu khác (SWOG). Phân tích theo kế hoạch đối với cả OS và PFS từ các bộ dữ liệu kết hợp của hai nghiên cứu này đã được tiến hành. Kết quả của sự phân tích kết hợp này được trình bày trong bảng 6.
- xem Bảng 6.

Trung vị thời gian theo dõi của các nghiên cứu kết hợp là 37,5 tháng (từ 25-75% là từ 19 đến 46 tháng). Có một cải thiện đáng kể có ý nghĩa thống kê về PFS trong nhóm điều trị 800 mg (23,2 so với 18,9 tháng) ở nhóm điều trị 400 mg (p=0,03). Tuy nhiên, không có sự khác biệt quan sát được về OS giữa các nhóm điều trị (p=0,98). PFS chung được ước tính cho tất cả 1640 bệnh nhân trong các nghiên cứu giai đoạn III này là 21 tháng và OS được ước tính là 48,8 tháng. Chỉ có 5,1% số bệnh nhân đạt được đáp ứng hoàn toàn được xác nhận và 47,5% đạt được đáp ứng một phần. Việc điều trị ở cả hai mức liều nhìn chung đều được hấp thu tốt và tất cả có 5,4% số bệnh nhân rút khỏi nghiên cứu do độc tính.
Một nghiên cứu giai đoạn II, đa quốc gia, ngẫu nhiên, nhãn mở được tiến hành trên bệnh nhân bị GIST ác tính không thể cắt bỏ hoặc đã di căn. Trong nghiên cứu này có 147 bệnh nhân tham gia và được chọn ngẫu nhiên dùng thuốc liều 400 mg hoặc 600 mg đường uống 1 lần/ngày cho đến 36 tháng.
Chứng cứ chủ yếu về hiệu quả dựa trên các tỷ lệ đáp ứng khách quan. Sự đánh giá về đáp ứng dựa trên tiêu chuẩn của Nhóm Nghiên cứu về Ung thư Tây Nam nước Mỹ (Southwestern Oncology Group - SWOG). Trong nghiên cứu này, 83% bệnh nhân hoặc có đáp ứng hoàn toàn, đáp ứng một phần hoặc là bệnh ổn định. Các kết quả được mô tả trong bảng 7.
- xem Bảng 7.

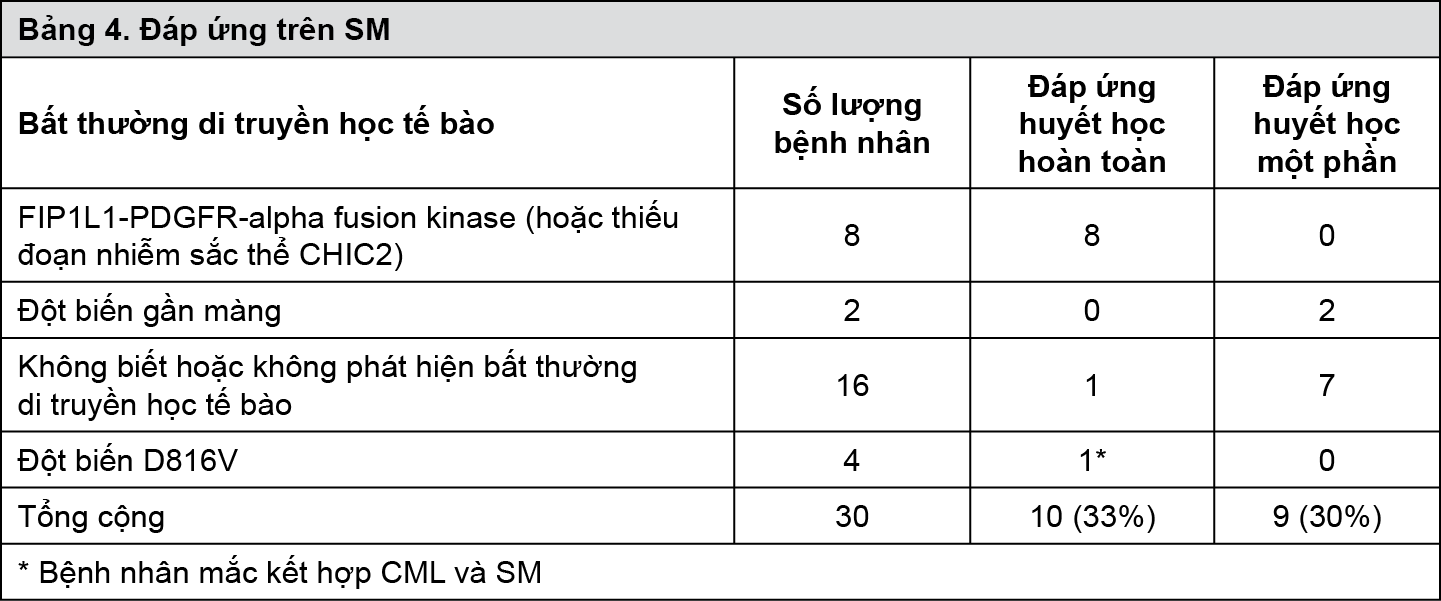
Không có sự khác nhau về tỷ lệ đáp ứng giữa 2 nhóm liều dùng (trung vị thời gian theo dõi là 31 tháng). Trung vị thời gian đến lúc đáp ứng là 13 tuần. Trung vị thời gian đến thất bại điều trị ở những người đáp ứng là 122 tuần, trong khi trong dân số nghiên cứu chung thời gian này là 84 tuần. Trung vị thời gian sống còn toàn bộ chưa ghi nhận được. Ước tính Kaplan-Meier về tỷ lệ sống còn sau thời gian theo dõi 36 tháng là 68% (Hình 2). Ngoài ra, không có sự khác biệt về tỷ lệ sống còn giữa bệnh nhân đạt được bệnh ổn định và bệnh nhân đáp ứng một phần (Hình 3).
- xem Hình 2 & Hình 3.

Các nghiên cứu lâm sàng trên điều trị bổ trợ cho GIST
Trong điều trị bổ trợ, Glivec được nghiên cứu trong một thử nghiệm giai đoạn III đa trung tâm, mù đôi, thời gian dài, có đối chứng với giả dược (Z9001) bao gồm 713 bệnh nhân.
Kết cục chính của nghiên cứu này là tỉ lệ sống không tái phát bệnh (RFS) được xác định là thời gian từ ngày bắt đầu được chọn ngẫu nhiên cho đến ngày bị tái phát bệnh hoặc tử vong vì bất cứ nguyên nhân gì.
Glivec đã kéo dài một cách đáng kể RFS với 75% số bệnh nhân không bị tái phát bệnh cho đến 38 tháng trong nhóm dùng Glivec so với 20 tháng trong nhóm dùng giả dược (KTC 95%, [30 bệnh nhân không đánh giá được ở nhóm dùng Glivec và 14 bệnh nhân không đánh giá được ở nhóm dùng giả dược]); (tỉ số nguy hại = 0,398 [0,259 đến 0,610], p<0,0001). Sau một năm RFS nói chung tốt hơn đáng kể ở nhóm dùng Glivec (97,7%) so với nhóm dùng giả dược (82,3%), (p<0,0001) do đó làm giảm nguy cơ tái phát bệnh khoảng 89% so với giả dược (tỉ số nguy hại = 0,113 [0,049 đến 0,264]).
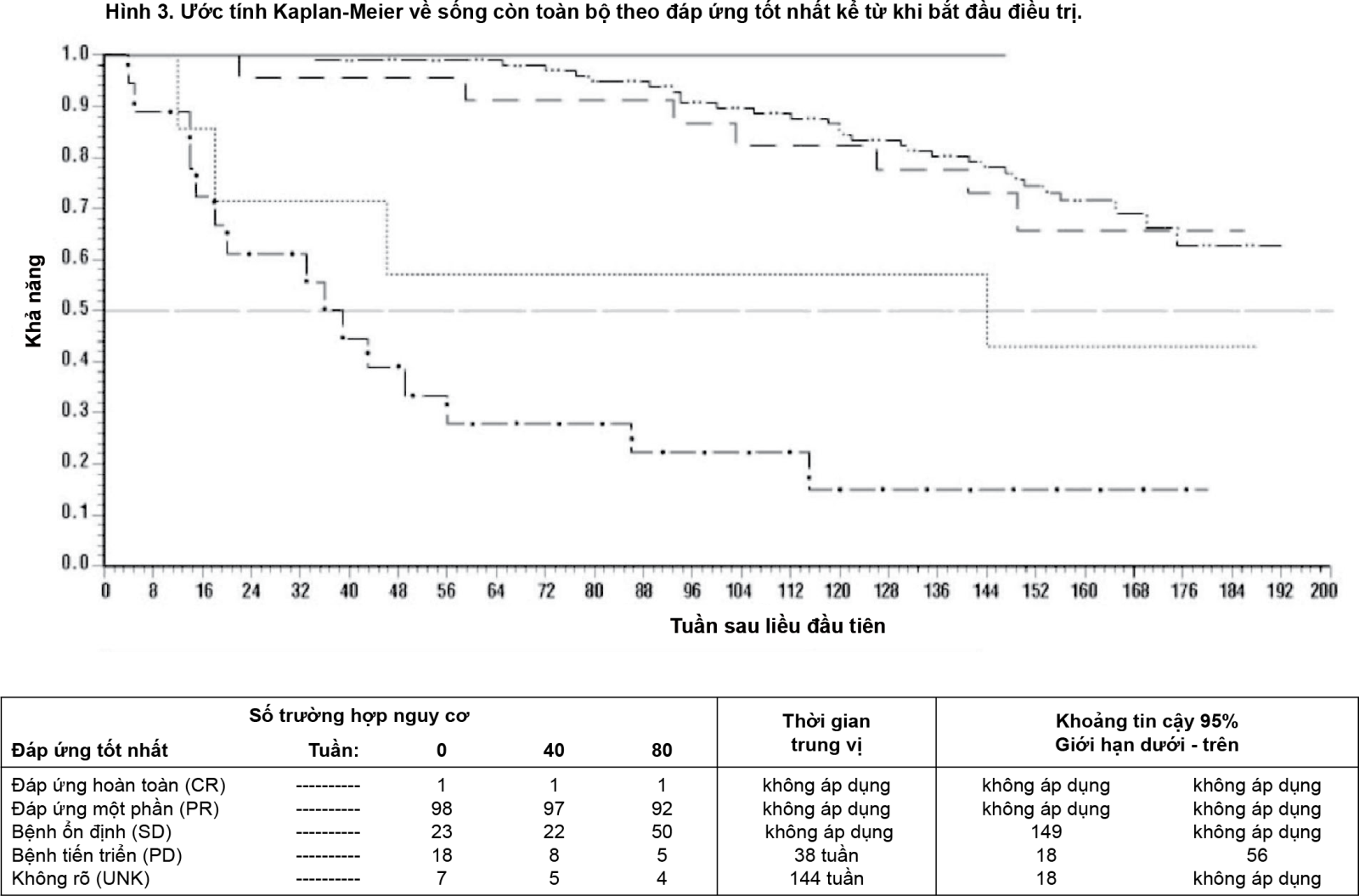
Một nghiên cứu nhãn mở pha III thứ hai (SSG XVIII/AIO) so sánh việc điều trị bằng Glivec 400 mg/ngày trong 12 tháng so với điều trị trong 36 tháng ở bệnh nhân sau phẫu thuật cắt bỏ GIST và một trong số sau: đường kính khối u > 5cm và chỉ số phân bào >5/50 trong quang trường có độ phóng đại cao (HPF); hoặc đường kính khối u > 10cm và bất kỳ chỉ số phân bào hoặc khối u với kích thước bất kỳ có chỉ số phân bào >10/50 HPF hoặc khối u vỡ trong khoang phúc mạc. Tổng số 397 bệnh nhân đồng ý và được chọn ngẫu nhiên để nghiên cứu (1 nhóm gồm 199 bệnh nhân điều trị 12 tháng và 1 nhóm gồm 198 bệnh nhân điều trị trong 36 tháng), tuổi trung vị là 61 (khoảng từ 22 đến 84 tuổi). Trung vị thời gian theo dõi là 54 tháng (từ ngày chọn ngẫu nhiên đến ngày kết dữ liệu), với tổng số 83 tháng từ lúc chọn ngẫu nhiên bệnh nhân đầu tiên đến kết dữ liệu.
Kết cục chính của nghiên cứu là thời gian sống còn không tái phát bệnh (Recurrence Free Survival - RFS) được định nghĩa là thời gian từ ngày chọn ngẫu nhiên đến ngày tái phát hoặc tử vong do bất cứ nguyên nhân nào. Ba mươi sáu (36) tháng điều trị Glivec kéo dài RFS đáng kể so với 12 tháng điều trị bằng Glivec (với Tỉ lệ nguy hại chung (Hazard Ratio – HR=0,46 [0,32; 0,65], p<0,0001 và HR=0,42 [0,28; 0,61] sau tháng 12) (bảng 8, Hình 4). Có tổng số 84 (42%) và 50 (25%) trường hợp RFS cho nhóm 12 tháng và 36 tháng.
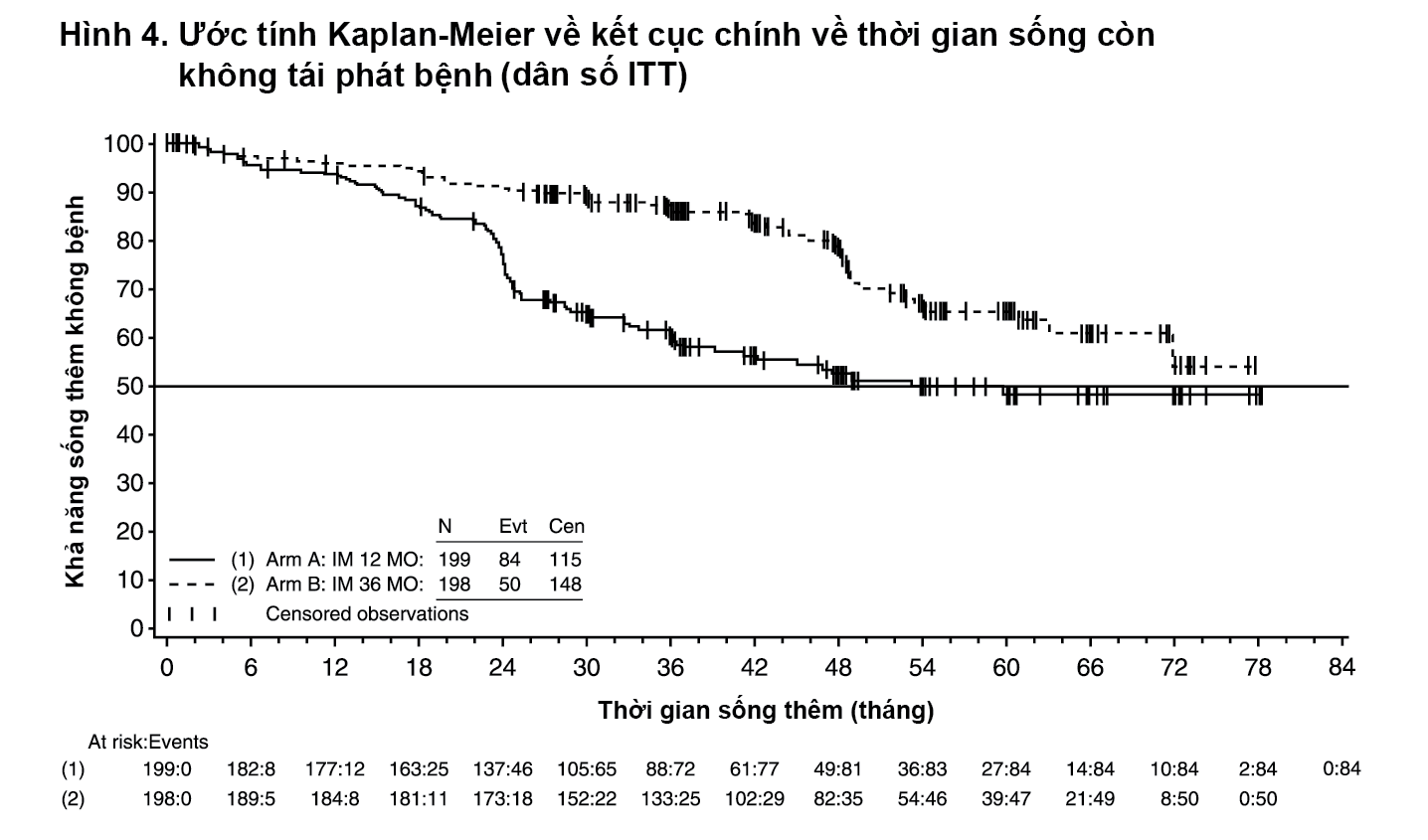
Ngoài ra, ba mươi sáu (36) tháng điều trị bằng Glivec kéo dài có ý nghĩa thời gian sống còn toàn bộ (Overall Survival – OS) so với nhóm điều trị bằng Glivec trong 12 tháng (HR=0,45 [0,22; 0,89], p=0,0187) (bảng 8, Hình 5). Tổng số tử vong là 25 trong nhóm điều trị 12 tháng và 12 trong nhóm điều trị 36 tháng.
- xem Bảng 8, Hình 4 & Hình 5.

Nghiên cứu lâm sàng trong bệnh DFSP: Một thử nghiệm lâm sàng giai đoạn II, đa quốc gia, nhãn mở (nghiên cứu B2225) đã được tiến hành dùng Glivec cho các nhóm bệnh nhân khác nhau bị các bệnh đe dọa tính mạng liên quan đến ABL, c-Kit hoặc PDGFR protein tyrosine kinase. Nghiên cứu này bao gồm 12 bệnh nhân bị u sarcom sợi bị lồi (DFSP) trong tổng số 185 bệnh nhân, 45 người trong số này bị các bệnh về máu và 140 người có khối u đặc các loại. Chứng cứ chính về hiệu quả đối với bệnh nhân ở nhóm khối u đặc dựa trên tỷ lệ đáp ứng khách quan. Nhóm khối u đặc được điều trị bằng Glivec 800 mg/ngày. Tuổi của các bệnh nhân bị DFSP từ 23 đến 75 tuổi, bệnh DFSP đã di căn, tái phát tại chỗ sau phẫu thuật cắt bỏ ban đầu và không được xem là có thể đáp ứng với phẫu thuật cắt bỏ thêm vào thời điểm nghiên cứu. Đã có báo cáo thêm 6 bệnh nhân bị DFSP được điều trị bằng Glivec trong 5 báo cáo ca bệnh đã được công bố, tuổi của các bệnh nhân này từ 18 tháng đến 49 tuổi. Tổng số trường hợp của nhóm được điều trị DFSP gồm 18 bệnh nhân, 8 trường hợp bệnh đã di căn. Những bệnh nhân người lớn được báo cáo trong y văn đã công bố được điều trị Glivec 400 mg/ngày (4 trường hợp) hoặc 800 mg/ngày (1 trường hợp). Bệnh nhân trẻ em được dùng 400 mg/m2/ngày sau đó tăng đến 520 mg/m2/ngày. Đáp ứng với điều trị được mô tả trong bảng 9.
- xem Bảng 9.

12 trong số 18 bệnh nhân này hoặc đã đạt được đáp ứng hoàn toàn (7 bệnh nhân) hoặc được điều trị hết bướu bằng phẫu thuật sau khi đáp ứng một phần (5 bệnh nhân bao gồm 1 trẻ em) tương ứng tỷ lệ đáp ứng hoàn toàn là 67%. Thêm 3 bệnh nhân đã đạt được đáp ứng một phần đối với tỷ lệ đáp ứng toàn bộ là 83%. Trong số 8 bệnh nhân có bệnh đã di căn, 5 người đã đáp ứng (62%), 3 người đáp ứng hoàn toàn (37%). Trung vị thời gian điều trị trong nghiên cứu B2225 là 6,2 tháng, với thời gian tối đa là 24,3 tháng, trong khi trên y văn đã công bố thời gian này khoảng từ 4 tuần đến hơn 20 tháng.
Nghiên cứu lâm sàng trong suy giảm chức năng gan: Trong một nghiên cứu trên bệnh nhân có các mức độ rối loạn chức năng gan khác nhau (nhẹ, trung bình và nặng - xem bảng 10 về xếp loại chức năng gan), nồng độ imatinib trung bình (diện tích dưới đường cong nồng độ (AUC) ở liều bình thường) không tăng so với bệnh nhân có chức năng gan bình thường. Trong nghiên cứu này, liều 500 mg/ngày là an toàn để sử dụng cho bệnh nhân rối loạn chức năng gan nhẹ và 300 mg dùng cho các bệnh nhân khác. Mặc dù chỉ có liều 300 mg/ngày được sử dụng cho bệnh nhân bị rối loạn chức năng gan trung bình và nặng, phân tích dược động học cho thấy 400 mg có thể được sử dụng an toàn (xem phần Liều lượng và Cách dùng, Cảnh báo, Tác dụng ngoại ý và Dược động học).
- xem Bảng 10.

Nghiên cứu lâm sàng trong suy thận: Trong một nghiên cứu trên bệnh nhân có các mức độ rối loạn chức năng thận khác nhau (nhẹ, trung bình và nặng - xem bảng 11 về xếp loại chức năng thận), nồng độ imatinib trung bình (diện tích dưới đường cong nồng độ ở liều bình thường) tăng 1,5-2 lần so với bệnh nhân có chức năng thận bình thường, tương ứng với tăng nồng độ trong huyết tương của AGP - một protein gắn mạnh vào imatinib. Chưa quan sát thấy có sự tương quan giữa nồng độ imatinib và độ trầm trọng của suy chức năng thận. Trong nghiên cứu này liều 800 mg/ngày là an toàn để sử dụng cho bệnh nhân rối loạn chức năng thận nhẹ và 600 mg/ngày dùng trong trường hợp rối loạn chức năng thận trung bình. Liều 800 mg không được thử nghiệm trên bệnh nhân bị rối loạn chức năng thận trung bình do số lượng bệnh nhân tham gia nghiên cứu còn giới hạn. Tương tự, chỉ có 2 bệnh nhân bị rối loạn chức năng thận nặng tham gia với liều thấp (100 mg), và không có liều cao nào được thử nghiệm. Không có bệnh nhân đang thẩm phân máu tham gia vào nghiên cứu. Các dữ liệu trong y văn cho thấy là liều 400 mg/ngày được dung nạp tốt ở bệnh nhân bị bệnh thận giai đoạn cuối đang thẩm phân máu. Nồng độ huyết tương về PK ở bệnh nhân này giảm xuống trong mức các trị số của imatinib và chất chuyển hóa của nó là CGP74588 quan sát thấy ở bệnh nhân có chức năng thận bình thường. Không thấy thẩm phân máu làm cản trở động học của imatinib trong huyết tương. Vì sự bài tiết qua thận là đường bài tiết thứ yếu của imatinib, bệnh nhân bị suy chức năng thận nặng và đang thẩm phân máu có thể được điều trị liều khởi đầu là 400 mg. Tuy nhiên, khuyến cáo thận trọng ở những bệnh nhân này. Có thể giảm liều này nếu không được dung nạp, hoặc tăng liều nếu thiếu hiệu quả (xem phần Liều lượng và Cách dùng, Cảnh báo, Tác dụng ngoại ý và Dược động học).
- xem Bảng 11.

Dược động học
Dược động học của Glivec được đánh giá ở mức liều 25-1.000 mg. Biểu đồ dược động học trong huyết tương được phân tích vào ngày thứ nhất và ngày thứ 7 hoặc ngày thứ 28 mà ở thời điểm đó nồng độ trong huyết tương đạt được trạng thái ổn định.
Hấp thu: Sinh khả dụng tuyệt đối trung bình đối với dạng bào chế viên nang imatinib là 98%. Hệ số biến thiên diện tích dưới đường cong nồng độ (AUC) của imatinib trong huyết tương ở mức 40% đến 60% sau khi dùng một liều uống. Khi ăn bữa ăn có nhiều chất béo, tỷ lệ hấp thu imatinib giảm tối thiểu (giảm 11% về nồng độ cao nhất trong huyết tương (Cmax) và kéo dài thời gian đạt nồng độ cao nhất trong huyết tương (tmax) đến 1,5 giờ), giảm nhẹ diện tích dưới đường cong nồng độ (7,4%) so với lúc đói.
Phân bố: Ở các nồng độ imatinib trên lâm sàng, tỷ lệ gắn với protein huyết tương khoảng 95% trên cơ sở các thực nghiệm in vitro, hầu hết gắn với albumin và alpha-acid-glycoprotein, gắn ít với lipoprotein.
Chuyển hóa: Chất chuyển hóa chính lưu thông ở người là chất dẫn xuất piperazine N-khử methyl (CGP71588) mà in vitro cho thấy hiệu lực tương tự như chất gốc. Diện tích dưới đường cong nồng độ trong huyết tương đối của chất chuyển hóa này chỉ bằng 16% diện tích dưới đường cong nồng độ của imatinib. Sự gắn với protein huyết tương của chất chuyển hóa N-khử methyl tương tự như chất gốc.
Thải trừ: Dựa trên việc tìm lại được các hợp chất sau khi uống một liều imatinib có gắn phóng xạ 14C, khoảng 81% liều được thải trừ trong vòng 7 ngày trong phân (68% liều) và nước tiểu (13% liều). Lượng imatinib không đổi chiếm 25% liều (5% trong nước tiểu, 20% trong phân), phần còn lại là các chất chuyển hóa.
Dược động học trong huyết tương: Sau khi cho những người tình nguyện khỏe mạnh dùng thuốc đường uống, thời gian bán thải (t½) khoảng 18 giờ, cho thấy rằng liều 1 lần/ngày là phù hợp. Sự tăng diện tích dưới đường cong nồng độ trung bình là tuyến tính với việc tăng liều và tỷ lệ với liều trong khoảng liều 25-1000 mg imatinib sau khi dùng đường uống. Không có sự thay đổi về động học của imatinib khi dùng liều lặp lại, và sự tích lũy gấp 1,5-2,5 lần ở trạng thái ổn định khi dùng liều 1 lần/ngày.
Dược động học trên nhóm dân số nghiên cứu
Dựa trên phân tích dược động học trên nhóm dân số nghiên cứu, tuổi có ít ảnh hưởng lên thể tích phân bố (tăng 12% ở bệnh nhân >65 tuổi). Thay đổi này không được cho là có ý nghĩa lâm sàng. Ảnh hưởng của thể trọng trên hệ số thanh thải imatinib trong trường hợp một bệnh nhân cân nặng 50kg, hệ số thanh thải trung bình sẽ là 8,5 L/giờ, trong khi một bệnh nhân cân nặng 100kg thì hệ số thanh thải sẽ đến 11,8 L/giờ. Những thay đổi này không được xem là đủ để bảo đảm cho việc điều chỉnh liều dựa trên kg thể trọng. Không có ảnh hưởng của giới tính đối với động học của imatinib.
Phân tích thêm về PK trên dân số nghiên cứu trong nghiên cứu giai đoạn III trên bệnh nhân bệnh bạch cầu tủy mạn mới được chẩn đoán cho thấy là tác dụng của các hiệp biến và phối hợp thuốc trên cả hệ số thanh thải và thể tích phân bố dường như là nhỏ và không đủ rõ để bảo đảm cho việc điều chỉnh liều.
Dược động học trên trẻ em
Giống như ở bệnh nhân người lớn, imatinib được hấp thu nhanh sau khi dùng đường uống đối với bệnh nhi ở cả 2 nghiên cứu giai đoạn I và giai đoạn II. Liều dùng cho trẻ em 260 mg/m2 đã đạt được nồng độ thuốc như khi dùng liều 400 mg ở người lớn, và liều 340 mg/m2 đã đạt được nồng độ thuốc như khi dùng liều 600 mg ở người lớn. Khi so sánh diện tích dưới đường cong nồng độ AUC(0-24) vào ngày thứ 8 và ngày thứ nhất với mức liều 340 mg/m2 đã phát hiện sự tích lũy thuốc gấp 1,7 lần sau khi dùng liều lặp lại 1 lần/ngày.
Dựa trên phân tích gộp dược động học trên bệnh nhân trẻ em với các rối loạn huyết học (CML, Ph+ALL, hoặc các rối loạn huyết học khác được điều trị bằng imatinib), độ thanh thải của imatinib tăng theo diện tích bề mặt cơ thể (BSA). Sau khi điều chỉnh cho ảnh hưởng của BSA, các yếu tố nhân khẩu học khác như là tuổi, trọng lượng cơ thể và chỉ số khối cơ thể không có ảnh hưởng có ý nghĩa lâm sàng lên nồng độ của imatnib. Phân tích đã khẳng định rằng nồng độ của imatinib ở bệnh nhân trẻ em dùng 260 mg/m2, 1 lần/ngày (không vượt quá 400 mg, 1 lần/ngày) hoặc 340 mg/m2, 1 lần/ngày (không vượt quá 600 mg, 1 lần/ngày) tương tự như bệnh nhân người lớn dùng 400 mg hoặc 600 mg, 1 lần/ngày.
Suy chức năng cơ quan
Imatinib và các chất chuyển hóa của nó không được bài tiết qua thận ở một mức độ đáng kể. Những bệnh nhân bị suy thận nhẹ và trung bình dường như có nồng độ thuốc trong huyết tương cao hơn so với bệnh nhân có chức năng thận bình thường. Sự gia tăng này gấp khoảng 1,5-2 lần, tương ứng với tăng 1,5 lần nồng độ AGP trong huyết tương là chất mà imatinib gắn kết mạnh. Hệ số thanh thải imatinib dạng tự do có lẽ giống nhau giữa bệnh nhân bị suy giảm chức năng thận và bệnh nhân có chức năng thận bình thường, vì sự bài tiết qua thận chỉ là đường thải trừ thứ yếu đối với imatinib (xem phần Liều lượng và Cách dùng, Cảnh báo và Dược lực học).
Mặc dù các kết quả phân tích dược động học cho thấy là có sự thay đổi đáng kể giữa các bệnh nhân, nồng độ imatinib trung bình không tăng ở bệnh nhân có các mức độ rối loạn chức năng gan khác nhau khi so sánh với bệnh nhân có chức năng gan bình thường (xem phần Liều lượng và Cách dùng, Cảnh báo, Tác dụng ngoại ý, Dược lực học và Dược động học).
THÔNG TIN TIỀN LÂM SÀNG
Imatinib đã được đánh giá trong các nghiên cứu về dược lý an toàn, độc tính liều lặp lại, độc tính gen và độc tính đối với sinh sản. Các cơ quan đích có liên quan với tác dụng dược lý của imatinib bao gồm tủy xương, máu ngoại vi, mô lymphô, tuyến sinh dục và đường tiêu hóa. Các cơ quan đích khác bao gồm gan và thận.
Imatinib có độc tính đối với phôi và gây quái thai ở chuột cống. Khả năng sinh sản không bị ảnh hưởng trong nghiên cứu tiền lâm sàng về khả năng sinh sản và nghiên cứu về sự phát triển phôi thai giai đoạn sớm mặc dù trọng lượng các tinh hoàn và mào tinh thấp hơn cũng như giảm số lượng tinh trùng có thể chuyển động đã được quan sát thấy ở những chuột đực dùng liều cao. Trong các nghiên cứu tiền lâm sàng trước và sau sinh trên chuột, khả năng sinh sản ở thế hệ con đầu tiên cũng không bị ảnh hưởng bởi Glivec.
Không nhận thấy có cơ quan đích mới trong nghiên cứu độc tính phát triển ở chuột chưa trưởng thành (ngày thứ 10 đến ngày 70 sau sinh). Trong nghiên cứu độc tính giai đoạn chưa trưởng thành, đã quan sát thấy các ảnh hưởng nhất thời trên sự tăng trưởng và trì hoãn sự mở lỗ âm đạo và sự tách nếp bao quy đầu khoảng 0,3 đến 2 lần nồng độ trung bình ở bệnh nhi khi dùng liều cao nhất được khuyến cáo là 340 mg/m2. Tương tự thế, tử vong được ghi nhận ở thú vật chưa trưởng thành (giai đoạn cai sữa) ở nồng độ khoảng 2 lần nồng độ trung bình ở bệnh nhi khi dùng liều cao nhất được khuyến cáo là 340 mg/m2.
Trong một nghiên cứu 2 năm về tính gây ung thư trên chuột cống dùng imatinib liều 15, 30 và 60 mg/kg/ngày đã dẫn đến giảm có ý nghĩa thống kê về thời gian sống của chuột đực ở liều 60 mg/kg/ngày và chuột cái ở liều ≥30 mg/kg/ngày. Khám nghiệm mô bệnh học chuột đã chết phát hiện bệnh cơ tim (cả hai giống), bệnh thận tiến triển mạn tính (chuột cái) và u nhú tuyến bao quy đầu là các nguyên nhân chính gây tử vong hoặc là lý do giết chuột để nghiên cứu. Các cơ quan đích có những thay đổi tân sinh là thận, bàng quang, niệu đạo, tuyến bao quy đầu và tuyến âm vật, ruột non, tuyến cận giáp, tuyến thượng thận và dạ dày không có tế bào tuyến. Các mức liều chưa quan sát thấy tác động (NOEL) đối với các cơ quan đích khác nhau về các tổn thương u tân sinh đã được xác định như sau: 30 mg/kg/ngày đối với thận, bàng quang, niệu đạo, ruột non, tuyến cận giáp, tuyến thượng thận và dạ dày không có tế bào tuyến, và 15 mg/kg/ngày đối với tuyến bao quy đầu và tuyến âm vật.
U nhú/ung thư biểu mô tuyến bao quy đầu/âm vật đã được ghi nhận ở liều 30 và 60 mg/kg/ngày, tương ứng với khoảng 0,5 đến 4 lần nồng độ hàng ngày ở người (dựa trên diện tích dưới đường cong nồng độ) khi dùng liều 400 mg/ngày hoặc khoảng 0,3 đến 2,4 lần nồng độ thuốc hàng ngày ở trẻ em (dựa trên diện tích dưới đường cong nồng độ) khi dùng liều 340 mg/m2. U tuyến thận/ung thư thận, u nhú bàng quang và niệu đạo, ung thư tuyến ruột non, u tuyến cận giáp, u tủy tuyến thượng thận lành tính và ác tính và ung thư biểu mô/u nhú dạ dày không có tế bào tuyến đã được ghi nhận khi dùng liều 60 mg/kg/ngày.
Chưa rõ có sự liên quan của các phát hiện này trong nghiên cứu về tính gây ung thư ở chuột cống đối với người. Phân tích các dữ liệu về độ an toàn từ các thử nghiệm lâm sàng và các báo cáo phản ứng phụ tự phát không thấy dấu hiệu tăng tỷ lệ ác tính nói chung ở bệnh nhân được điều trị bằng Glivec so với dân số nói chung.
Các tổn thương không phải u tân sinh không được ghi nhận trong các nghiên cứu tiền lâm sàng sớm là hệ tim mạch, tụy, cơ quan nội tiết và răng. Những thay đổi quan trọng nhất bao gồm phì đại tim và giãn tim, dẫn đến dấu hiệu suy tim ở một số động vật.
Chỉ định/Công dụng
Glivec được chỉ định để:
• điều trị bệnh nhân người lớn và trẻ em trên 2 tuổi bị bệnh bạch cầu tủy mạn với nhiễm sắc thể Philadelphia dương tính (Ph + CML) mới được chẩn đoán (để sử dụng cho bệnh nhi xin xem phần Liều lượng và Cách dùng).
• điều trị bệnh nhân người lớn và trẻ em trên 2 tuổi bị bệnh bạch cầu tủy mạn với nhiễm sắc thể Philadelphia dương tính (Ph + CML) trong cơn nguyên bào, giai đoạn tăng tốc hoặc giai đoạn mạn tính sau khi đã thất bại với điều trị interferon-alpha (để sử dụng cho bệnh nhi xin xem phần Liều lượng và Cách dùng).
• điều trị phối hợp với hóa trị liệu cho bệnh nhân người lớn và trẻ em bị bệnh bạch cầu nguyên bào lymphô cấp với nhiễm sắc thể Philadelphia dương tính (Ph+ ALL) mới được chẩn đoán.
• điều trị bệnh nhân người lớn bị bệnh bạch cầu nguyên bào lymphô cấp với nhiễm sắc thể Philadelphia dương tính tái phát hoặc kháng lại khi dùng đơn trị liệu.
• điều trị bệnh nhân người lớn bị bệnh loạn sản tủy/rối loạn tăng sinh tủy xương (MDS/MPD) có liên quan tới sự sắp xếp lại gen của thụ thể yếu tố tăng trưởng có nguồn gốc tiểu cầu (PDGFR).
• điều trị bệnh nhân người lớn bị bệnh lý dưỡng bào hệ thống (SM) không có đột biến D816V c-Kit hoặc chưa biết tình trạng đột biến c-Kit.
• điều trị bệnh nhân người lớn bị hội chứng tăng bạch cầu ái toan (HES) và/hoặc bệnh bạch cầu mạn tế bào ái toan (CEL).
• điều trị bệnh nhân người lớn bị u mô đệm dạ dày ruột (GIST) ác tính với c-Kit + (CD117) không thể cắt bỏ và/hoặc đã di căn.
• điều trị bổ trợ cho bệnh nhân người lớn sau phẫu thuật cắt bỏ GIST c-Kit+.
• điều trị bệnh nhân người lớn sarcom sợi bì lồi (DFSP) không thể cắt bỏ, tái phát và/hoặc đã di căn.
Liều lượng & Cách dùng
Việc điều trị nên được bắt đầu khi thích hợp bởi một bác sĩ có kinh nghiệm trong việc điều trị bệnh nhân bị các u ác tính về huyết học và các sarcom ác tính.
Nên uống liều được kê đơn cùng với bữa ăn và một ly nhiều nước để giảm thiểu nguy cơ rối loạn tiêu hóa. Liều 400 mg hoặc 600 mg nên dùng một lần/ngày trong khi liều 800 mg/ngày nên dùng viên 400mg, 2 lần/ngày, vào buổi sáng và buổi tối.
Đối với những bệnh nhân không thể nuốt viên nén bao phim, có thể hòa viên nén vào một ly nước hoặc nước táo. Số viên cần dùng nên được cho vào một lượng thức uống thích hợp (khoảng 50mL cho 1 viên 100mg và 200mL cho 1 viên 400mg) và khuấy bằng muỗng. Hỗn dịch phải được dùng ngay sau khi viên nén đã tan rã hoàn toàn.
Cần tiếp tục điều trị chừng nào vẫn còn có lợi cho bệnh nhân.
Theo dõi đáp ứng với Glivec ở những bệnh nhân Ph+CML nên được tiến hành thường xuyên và khi có điều chỉnh liệu pháp điều trị, để xác định đáp ứng dưới mức tối ưu, việc mất đáp ứng với điều trị, việc bệnh nhân tuân thủ kém, hoặc tương tác thuốc-thuốc có thể xảy ra. Kết quả theo dõi sẽ hướng dẫn cách xử trí CML thích hợp.
Đối tượng bệnh nhân thông thường
Liều dùng đối với bệnh CML
Liều Glivec khuyến cáo là 400 mg/ngày đối với bệnh nhân người lớn bị bệnh CML giai đoạn mạn tính và 600 mg/ngày đối với bệnh nhân trong giai đoạn tăng tốc hoặc cơn nguyên bào.
Có thể xem xét tăng liều từ 400 mg đến 600 mg hoặc 800 mg ở bệnh nhân bị bệnh giai đoạn mạn tính, hoặc từ 600 mg đến tối đa 800 mg/ngày ở bệnh nhân trong giai đoạn tăng tốc hoặc cơn nguyên bào khi không có phản ứng phụ nặng với thuốc và giảm bạch cầu trung tính hoặc giảm tiểu cầu nặng không liên quan đến bệnh bạch cầu trong những tình trạng sau đây: tiến triển bệnh (ở bất kỳ thời điểm nào), không đạt được đáp ứng huyết học thỏa đáng sau ít nhất 3 tháng điều trị, không đạt được đáp ứng di truyền học tế bào sau 12 tháng điều trị, hoặc mất đáp ứng về huyết học và/hoặc di truyền học tế bào đã đạt được trước đây.
Liều dùng cho trẻ em trên 2 tuổi: Liều dùng cho trẻ em nên dựa trên diện tích bề mặt cơ thể (mg/m2). Liều 340 mg/m2/ngày được khuyến cáo cho trẻ em bị CML giai đoạn mạn tính và giai đoạn tiến triển (không được vượt quá tổng liều 600 mg/ngày). Có thể điều trị một liều 1 lần/ngày hoặc một cách khác là liều hàng ngày có thể chia làm 2 lần dùng - một lần vào buổi sáng và một lần vào buổi tối (xem phần Dược lý).
Liều dùng đối với bệnh Ph+ ALL
Liều Glivec khuyến cáo là 600 mg/ngày cho bệnh nhân người lớn bị Ph+ ALL.
Liều dùng cho trẻ em: Liều dùng cho trẻ em nên dựa trên diện tích bề mặt cơ thể (mg/m2). Liều 340 mg/m2/ngày được khuyến cáo cho trẻ em bị Ph+ ALL (không được vượt quá tổng liều 600 mg/ngày). Có thể điều trị một liều 1 lần/ngày.
Liều dùng đối với bệnh MDS/MPD: Liều Glivec khuyến cáo là 400 mg/ngày cho bệnh nhân người lớn bị MDS/MPD.
Liều dùng đối với bệnh SM
Liều Glivec khuyến cáo là 400 mg/ngày cho bệnh nhân người lớn bị SM không có đột biến D816V c-Kit hoặc tình trạng đột biến không rõ hoặc không đáp ứng đầy đủ với các trị liệu khác.
Đối với bệnh nhân bị SM kết hợp với tăng bạch cầu ưa eosin, một bệnh huyết học theo dòng có liên quan đến fusion kinase FIP1L1-PDGFR-alpha, liều khởi đầu 100 mg/ngày được khuyến cáo. Việc tăng liều từ 100 mg lên 400 mg cho những bệnh nhân này có thể được xem xét khi không bị các tác dụng phụ của thuốc, nếu việc đánh giá cho thấy rằng đáp ứng với điều trị là chưa đầy đủ.
Liều dùng đối với bệnh HES/CEL
Liều Glivec khuyến cáo là 400 mg/ngày cho bệnh nhân người lớn bị HES/CEL.
Đối với bệnh nhân bị HES/CEL với FIP1L1-PDGFR-alpha fusion kinase đã được chứng minh, liều khởi đầu 100 mg/ngày được khuyến cáo. Việc tăng liều từ 100 mg lên 400 mg cho những bệnh nhân này có thể được xem xét khi không bị các tác dụng phụ của thuốc, nếu việc đánh giá cho thấy rằng đáp ứng với điều trị là chưa đầy đủ.
Liều dùng đối với bệnh GIST
Liều Glivec khuyến cáo là 400 mg/ngày cho bệnh nhân người lớn bị GIST ác tính không thể cắt bỏ và/hoặc đã di căn.
Có thể xem xét tăng liều từ 400 mg đến 600 mg hoặc 800 mg cho bệnh nhân khi không có các phản ứng phụ với thuốc nếu đánh giá thấy đáp ứng điều trị không đầy đủ.
Liều Glivec khuyến cáo là 400 mg/ngày để điều trị bổ trợ cho bệnh nhân người lớn sau phẫu thuật cắt bỏ GIST. Thời gian điều trị tối thiểu được khuyến cáo là 36 tháng.
Thời gian điều trị tối ưu với Glivec cho điều trị bổ trợ chưa được rõ.
Liều dùng đối với bệnh DFSP: Liều Glivec khuyến cáo là 800 mg/ngày cho bệnh nhân người lớn bị DFSP.
Điều chỉnh liều đối với các phản ứng phụ của thuốc
Phản ứng phụ của thuốc không thuộc huyết học
Nếu xảy ra một phản ứng phụ của thuốc không thuộc huyết học ở mức độ nặng khi dùng Glivec, phải ngừng điều trị cho đến khi biến cố này được giải quyết. Sau đó có thể điều trị trở lại khi thích hợp tùy thuộc vào độ nặng ban đầu của phản ứng phụ đó.
Nếu nồng độ bilirubin tăng hơn 3 lần giới hạn trên của mức bình thường (IULN) hoặc transaminase gan tăng hơn 5 lần IULN, phải ngừng Glivec cho đến khi nồng độ bilirubin trở về <1,5 lần IULN và nồng độ transaminase trở về <2,5 lần IULN. Sau đó có thể tiếp tục điều trị Glivec với liều hàng ngày đã được giảm. Ở người lớn, nên giảm liều từ 400 mg xuống 300 mg, hoặc từ 600 mg xuống 400 mg, hoặc từ 800 mg xuống 600 mg, và đối với trẻ em từ 340 mg/m2/ngày xuống 260 mg/m2/ngày.
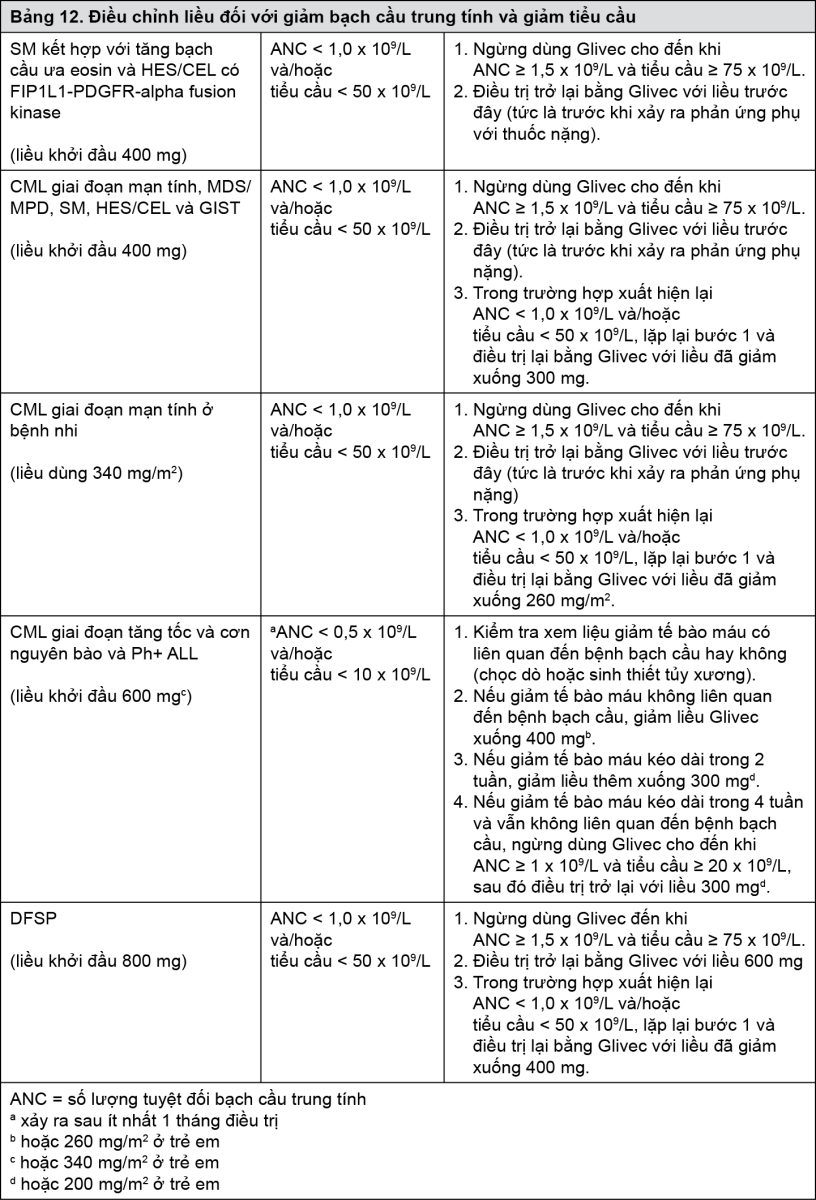
Phản ứng phụ của thuốc về huyết học: Khuyến cáo giảm liều hoặc ngừng điều trị đối với giảm bạch cầu trung tính và giảm tiểu cầu nặng được biểu thị trong bảng 12.
- xem Bảng 12.
Đối tượng đặc biệt
Trẻ em
Chưa có kinh nghiệm về việc sử dụng Glivec cho trẻ em dưới 2 tuổi bị CML và trẻ em dưới 1 tuổi bị Ph+ ALL. Có rất ít đến không có kinh nghiệm về việc sử dụng Glivec cho trẻ em ở các chỉ định khác.
Liều dùng cho trẻ em nên dựa trên diện tích bề mặt cơ thể (mg/m2). Liều 340 mg/m2/ngày được khuyến cáo cho trẻ em bị CML và Ph+ ALL giai đoạn mạn tính và giai đoạn tiến triển (không được vượt quá tổng liều 600 mg/ngày). Có thể điều trị một liều 1 lần/ngày trong chỉ định CML và Ph+ ALL. Đối với chỉ định CML liều hàng ngày có thể chia làm 2 lần dùng - một lần vào buổi sáng và một lần vào buổi tối (xem phần Dược lý).
Suy gan: Imatinib được chuyển hóa chủ yếu bởi gan. Bệnh nhân bị rối loạn chức năng gan nhẹ, trung bình hoặc nặng nên dùng liều thấp nhất được khuyến cáo là 400 mg/ngày. Liều này có thể giảm xuống nếu không được dung nạp (xem phần Cảnh báo, Tác dụng ngoại ý và Dược lý).
Giảm chức năng thận: Imatinib và các chất chuyển hóa của nó được bài tiết không đáng kể qua thận. Những bệnh nhân bị rối loạn chức năng thận hoặc đang thẩm phân có thể dùng liều khởi đầu thấp nhất được khuyến cáo là 400 mg/ngày (xem phần Dược lý). Tuy nhiên, khuyến cáo thận trọng đối với những bệnh nhân này. Liều này có thể giảm xuống nếu không được dung nạp. Nếu được dung nạp, liều có thể tăng lên nếu thiếu hiệu quả (xem phần Cảnh báo).
Bệnh nhân cao tuổi: Không quan sát thấy sự khác biệt đáng kể về dược động học liên quan đến tuổi ở bệnh nhân người lớn trong các thử nghiệm lâm sàng bao gồm trên 20% bệnh nhân từ 65 tuổi trở lên. Không cần liều khuyến cáo đặc biệt cho bệnh nhân cao tuổi.
Cảnh báo
Thuốc này chỉ dùng theo sự kê đơn của bác sĩ.
Khi dùng phối hợp Glivec với các thuốc khác, có khả năng xảy ra tương tác thuốc. Cần thận trọng khi sử dụng Glivec cùng với rifampicin hoặc các thuốc kích thích mạnh CYP3A4 khác, ketoconazole hoặc các thuốc ức chế mạnh CYP3A4, cơ chất của CYP3A4 có cửa sổ điều trị hẹp (ví dụ cyclosporin hoặc pimozide) hoặc cơ chất CYP2C9 có cửa sổ điều trị hẹp (ví dụ warfarin và dẫn chất coumarin khác) (xem phần Tương tác).
Suy giáp: Đã có báo cáo về các trường hợp suy giáp trên lâm sàng ở bệnh nhân cắt tuyến giáp đang dùng levothyroxine thay thế trong khi điều trị bằng Glivec. Cần theo dõi chặt chẽ nồng độ hormon kích thích tuyến giáp (TSH) ở những bệnh nhân này.
Nhiễm độc gan: Ở bệnh nhân bị rối loạn chức năng gan (nhẹ, trung bình hoặc nặng), phải theo dõi chặt chẽ công thức máu ngoại vi và các enzyme gan (xem phần Liều lượng và Cách dùng, Tác dụng ngoại ý, Cơ chế tác dụng, Dược lý).
Khi Glivec được kết hợp với hóa liệu pháp liều cao, đã quan sát thấy độc tính gan thoáng qua dưới dạng tăng transaminase và tăng bilirubin máu. Hơn nữa, đã có những báo cáo không thường gặp về suy gan cấp. Khuyến cáo theo dõi chức năng gan trong trường hợp Glivec được kết hợp với các phác đồ hóa trị liệu đã biết là có liên quan với rối loạn chức năng gan (xem phần Tác dụng ngoại ý).
Ứ dịch: Đã có báo cáo về ứ dịch nặng (tràn dịch màng phổi, phù, phù phổi, cổ trướng và phù bề mặt) trong khoảng 2,5% bệnh nhân bị bệnh bạch cầu tủy mạn mới được chẩn đoán đang dùng Glivec. Vì vậy, bệnh nhân nên được cân đều đặn. Phải kiểm tra cẩn thận hiện tượng tăng cân nhanh bất thường và nếu cần nên tiến hành các biện pháp chăm sóc hỗ trợ và điều trị thích hợp. Trong các thử nghiệm lâm sàng, tỷ lệ các biến cố này tăng lên ở bệnh nhân cao tuổi và những người có tiền sử về bệnh tim.
Bệnh nhân bị bệnh tim hoặc suy thận: Bệnh nhân bị bệnh tim, có các yếu tố nguy cơ suy tim hoặc có tiền sử suy thận phải được kiểm soát chặt chẽ và bất cứ bệnh nhân nào có dấu hiệu hoặc triệu chứng phù hợp với suy tim hoặc suy thận đều phải được đánh giá và điều trị.
Ở những bệnh nhân bị hội chứng tăng bạch cầu ái toan (HES) với sự thâm nhiễm ẩn của tế bào HES vào trong cơ tim, các trường hợp riêng lẻ bị sốc tim/rối loạn chức năng thất trái có liên quan đến sự mất hạt tế bào HES khi khởi đầu điều trị bằng Glivec. Tình trạng này được báo cáo là phục hồi với điều trị bằng steroid toàn thân, các biện pháp hỗ trợ tuần hoàn và ngừng Glivec tạm thời. Bệnh loạn sản tủy (MDS)/rối loạn tăng sinh tủy xương (MPD) và u dưỡng bào hệ thống có thể kết hợp với các mức bạch cầu ái toan cao. Do đó làm siêu âm tim và xác định troponin huyết thanh phải được xem xét ở những bệnh nhân bị HES/CEL, và những bệnh nhân bị MDS/MPD hoặc SM kết hợp với các mức bạch cầu ái toan cao. Nếu có bất thường, nên xem xét việc dùng dự phòng steroid toàn thân (1-2 mg/kg) trong 1-2 tuần đồng thời với Glivec khi bắt đầu điều trị.
Xuất huyết tiêu hóa: Trong các nghiên cứu giai đoạn III về GIST trên bệnh nhân bị GIST ác tính không thể cắt bỏ hoặc đã di căn đã có 211 bệnh nhân (12,9%) được báo cáo có xuất huyết mức độ 3/4 ở vị trí bất kỳ. Trong nghiên cứu giai đoạn II trên các bệnh nhân bị GIST ác tính không thể cắt bỏ hoặc đã di căn (nghiên cứu B2222), tám bệnh nhân (5,4%) bị xuất huyết tiêu hóa và bốn bệnh nhân (2,7%) bị xuất huyết ở khối u. Tùy thuộc vào vị trí giải phẫu của thương tổn khối u mà xuất huyết khối u là xuất huyết trong bụng hoặc trong gan. Các vị trí trong đường tiêu hóa của khối u có thể góp phần vào các báo cáo về chảy máu đường tiêu hóa ở nhóm bệnh nhân này. Bên cạnh đó, giãn mạch máu hang vị (GAVE), một nguyên nhân hiếm gặp gây xuất huyết tiêu hóa, đã được báo cáo trong kinh nghiệm hậu mãi với các bệnh nhân CML, ALL và một số bệnh khác. Do đó các bệnh nhân cần được kiểm tra các triệu chứng của đường tiêu hóa khi bắt đầu và trong suốt quá trình điều trị bằng Glivec. Khi cần, có thể cân nhắc việc ngưng dùng Glivec (xem phần Tác dụng ngoại ý).
Hội chứng ly giải khối u: Các trường hợp hội chứng ly giải khối u (TLS) đã được báo cáo ở các bệnh nhân điều trị bằng Glivec. Do có thể xuất hiện TLS, khuyến cáo nên điều trị tình trạng mất nước có ý nghĩa lâm sàng và điều trị nồng độ acid uric cao trước khi bắt đầu điều trị bằng Glivec (xem phần Tác dụng ngoại ý).
Tái hoạt hóa viêm gan B: Sự tái hoạt hóa viêm gan B có thể xảy ra ở những bệnh nhân nhiễm vi rút này mạn tính sau khi sử dụng một chất ức chế tyrosin kinase (TKI) BCR-ABL, ví dụ như imatinib. Một vài trường hợp có liên quan đến việc sử dụng nhóm thuốc BCR-ABL TKI dẫn đến suy gan cấp hoặc viêm gan tối cấp dẫn đến phải ghép gan hoặc tử vong (xem phần Tác dụng ngoại ý).
Bệnh nhân nên được xét nghiệm phát hiện nhiễm vi rút viêm gan B trước khi khởi đầu điều trị bằng imatinib. Những bệnh nhân hiện tại đang sử dụng imatinib nên được xét nghiệm ban đầu về nhiễm vi rút viêm gan B để xác định những người nhiễm vi rút mạn tính. Nên hỏi ý kiến các chuyên gia về bệnh gan và điều trị viêm gan B trước khi khởi đầu điều trị ở bệnh nhân có huyết thanh dương tính với vi rút viêm gan B (kể cả những người bị viêm gan thể hoạt động) và bệnh nhân có xét nghiệm dương tính với vi rút viêm gan B trong quá trình trị liệu. Những người mang vi rút viêm gan B cần điều trị bằng imatinib nên được theo dõi chặt chẽ để phát hiện các triệu chứng và dấu hiệu của nhiễm vi rút viêm gan B thể hoạt động trong suốt quá trình trị liệu và vài tháng sau khi chấm dứt trị liệu.
Xét nghiệm
Phải tiến hành làm công thức máu toàn phần thường xuyên trong thời gian điều trị bằng Glivec. Việc điều trị bệnh nhân bị bệnh bạch cầu tủy mạn bằng Glivec có liên quan với giảm bạch cầu trung tính hoặc giảm tiểu cầu. Tuy nhiên, sự giảm tế bào máu này phụ thuộc vào giai đoạn bệnh được điều trị và thường gặp hơn ở những bệnh nhân bị bệnh bạch cầu tủy mạn giai đoạn tăng tốc hoặc cơn nguyên bào so với những bệnh nhân bị bệnh bạch cầu tủy mạn giai đoạn mạn tính. Có thể ngừng điều trị bằng Glivec hoặc giảm liều như đã khuyến cáo trong phần Liều lượng và Cách dùng.
Phải theo dõi chức năng gan đều đặn (transaminase, bilirubin, phosphatase kiềm) ở bệnh nhân đang dùng Glivec. Như đã khuyến cáo trong phần Liều lượng và Cách dùng, phần "Phản ứng phụ của thuốc không thuộc huyết học", những bất thường về xét nghiệm này phải được xử trí bằng cách ngừng dùng và/hoặc giảm liều điều trị Glivec.
Glivec và các chất chuyển hóa của nó không được bài tiết qua thận ở một mức độ đáng kể. Hệ số thanh thải Creatinin (CrCL) được biết là giảm theo tuổi, nhưng tuổi không ảnh hưởng đáng kể đến động học của Glivec. Ở bệnh nhân bị suy giảm chức năng thận, sự tồn lưu imatinib trong huyết tương dường như cao hơn so với bệnh nhân có chức năng thận bình thường, có lẽ là do nồng độ alpha-acid glycoprotein (AGP) trong huyết tương - là một protein gắn với imatinib, tăng lên ở những bệnh nhân này. Không có sự tương quan giữa nồng độ imatinib và mức độ suy giảm chức năng thận phân loại bằng cách đo hệ số thanh thải creatinin (CrCL), giữa các bệnh nhân bị suy giảm chức năng thận nhẹ (CrCL: 40-59ml/phút) và nặng (CrCL: < 20ml/phút). Tuy nhiên, như đã khuyến cáo trong phần Liều lượng và Cách dùng, có thể giảm liều Glivec khởi đầu nếu không được dung nạp.
Trẻ em và thiếu niên: Đã có các báo cáo ca bệnh về chậm tăng trưởng xảy ra ở trẻ em và thiếu niên dùng Glivec. Chưa biết rõ ảnh hưởng lâu dài khi điều trị dài ngày bằng Glivec trên tăng trưởng của trẻ em. Vì thế, khuyến cáo nên theo dõi chặt chẽ sự tăng trưởng của trẻ em khi điều trị bằng Glivec (xem phần Tác dụng ngoại ý).
Ảnh hưởng của thuốc đối với công việc: Đã có các báo cáo về tai nạn khi lái xe ở các bệnh nhân dùng Glivec. Trong khi hầu hết các báo cáo này không bị nghi ngờ là gây ra bởi Glivec, bệnh nhân nên được khuyến cáo rằng họ có thể bị các tác dụng không mong muốn như là chóng mặt, nhìn mờ, hoặc ngủ gà trong khi dùng Glivec. Vì thế, cần thận trọng khi lái xe hoặc vận hành máy móc.
Quá Liều
Quá liều
Chưa có nhiều kinh nghiệm về việc sử dụng các liều cao hơn liều điều trị. Các trường hợp riêng lẻ về quá liều Glivec đã được báo cáo một cách tự phát và trong y văn. Nhìn chung kết cục của các trường hợp này là được báo cáo là có cải thiện hoặc hồi phục. Trong trường hợp quá liều, bệnh nhân phải được theo dõi và có các biện pháp điều trị triệu chứng thích hợp.
Các biến cố được báo cáo ở các khoảng liều khác nhau như sau:
Quá liều ở người lớn
Từ 1.200 đến 1.600 mg (với khoảng thời gian thay đổi giữa 1 đến 10 ngày): buồn nôn, nôn, tiêu chảy, phát ban, ban đỏ, phù, sưng tấy, mệt mỏi, co cứng cơ, giảm tiểu cầu, giảm toàn thể huyết cầu, đau bụng, đau đầu, giảm ngon miệng. Từ 1.800 đến 3.200 mg (tương đương với 3.200 mg/ngày trong 6 ngày): yếu ớt, đau cơ, tăng CPK, tăng bilirubin, đau dạ dày ruột. 6.400 mg (liều duy nhất): một trường hợp trong y văn được báo cáo, bệnh nhân bị buồn nôn, nôn, đau bụng, sốt, sưng tấy mặt, giảm lượng bạch cầu trung tính, tăng các transaminase.
Từ 8 đến 10 g (liều duy nhất): nôn và đau dạ dày ruột đã được báo cáo.
Quá liều ở trẻ em: Một bé trai 3 tuổi dùng liều duy nhất 400 mg bị nôn, tiêu chảy và chán ăn và một bé trai 3 tuổi khác dùng liều duy nhất 980 mg bị giảm lượng bạch cầu và tiêu chảy.
Cách xử lý: Tích cực theo dõi để có biện pháp xử trí kịp thời.
Chống chỉ định
Bệnh nhân quá mẫn cảm với hoạt chất hoặc với bất kỳ thành phần nào của tá dược.
Sử dụng ở phụ nữ có thai và cho con bú
Phụ nữ có khả năng mang thai: Phụ nữ có khả năng mang thai phải được khuyên dùng một biện pháp ngừa thai có hiệu quả cao trong thời gian điều trị Glivec. Biện pháp ngừa thai có hiệu quả cao là một phương pháp tránh thai cho tỉ lệ kết quả thất bại thấp (ví dụ ít hơn 1%/năm) khi sử dụng kiên định và đúng.
Có thai: Nghiên cứu trên động vật cho thấy độc tính trên hệ sinh sản (xem phần An toàn tiền lâm sàng). Không có thử nghiệm lâm sàng trên các phụ nữ có thai dùng Glivec. Đã có báo cáo hậu mãi về sẩy thai tự phát và dị tật bẩm sinh ở trẻ nhũ nhi trên phụ nữ dùng Glivec. Glivec chỉ nên được dùng trong thai kỳ khi lợi ích mong đợi vượt trội nguy cơ tiềm ẩn đối với thai nhi. Bệnh nhân phải được thông báo về các nguy cơ tiểm ẩn đối với thai nhi nếu thuốc được dùng trong thai kỳ.
Cho con bú: Cả imatinib và chất chuyển hóa có hoạt tính của nó đều có thể được bài tiết vào sữa mẹ. Tỉ lệ sữa huyết tương được xác định là 0,5 với imatinib và 0,9 với chất chuyển hóa, cho thấy chất chuyển hóa của imatinib được bài tiết vào sữa mẹ nhiều hơn. Xem xét về nồng độ kết hợp của imatinib và chất chuyển hóa và lượng sữa tối đa hàng ngày trẻ bú vào thì tổng lượng thuốc phơi nhiễm được coi là thấp (khoảng 10% liều điều trị). Tuy vậy, phụ nữ đang dùng Glivec không nên cho con bú do các tác động của việc phơi nhiễm imatinib ở liều thấp ở trẻ nhỏ chưa được biết đến.
Khả năng sinh sản: Các nghiên cứu trên bệnh nhân nam dùng Glivec và ảnh hưởng của nó đến khả năng sinh sản nam và sự sinh tinh chưa được thực hiện. Các bệnh nhân nam lo ngại về khả năng sinh sản của họ khi điều trị bằng Glivec nên tham khảo ý kiến bác sỹ (xem phần An toàn tiền lâm sàng).
Tương tác
Các tương tác quan sát được dẫn đến việc không khuyến cáo sử dụng đồng thời
Những thuốc có thể làm giảm nồng độ imatinib trong huyết tương: Những chất gây thúc đẩy hoạt động của CYP3A4 (ví dụ dexamethasone, phenytoin, carbamazepine, rifampicin, phenobarbital hoặc Hypericum perforatum – còn được biết là cỏ St. John’s Wort) có thể làm giảm đáng kể sự tồn lưu Glivec. Điều trị trước cho 14 người tình nguyện khỏe mạnh với nhiều liều rifampicin 600 mg/ngày trong vòng 8 ngày, sau đó dùng liều đơn Glivec 400 mg, sự thanh thải khi dùng Glivec liều uống tăng lên gấp 3,8 lần (90% khoảng tin cậy = gấp 3,5 đến 4,3 lần), điều này biểu thị sự giảm Cmax trung bình là 54%, AUC(0-24) là 68% và AUC(0-∞) là 74% khi không điều trị rifampicin. Các kết quả tương tự cũng được quan sát thấy ở những bệnh nhân bị u thần kinh đệm ác tính được điều trị với Glivec trong khi đang dùng các thuốc chống động kinh gây cảm ứng enzyme (EIAED) như carbamazepine, oxcarbazepine, phenytoin, fosphenytoin, phenobarbital và primidone. AUC huyết tương của imatinib giảm 73% so với những bệnh nhân không dùng EIAED. Trong hai nghiên cứu đã được công bố, dùng đồng thời Glivec và một sản phẩm chứa cỏ St. John’s Wort dẫn đến giảm 30 đến 32% AUC của Glivec. Ở những bệnh nhân được chỉ định dùng rifampicin hoặc các chất gây thúc đẩy CYP3A4 khác, nên xem xét thay đổi thuốc điều trị ít có khả năng gây cảm ứng enzyme.
Những tương tác khác có thể ảnh hưởng đến sự tồn lưu của Glivec hoặc các thuốc khác
Những thuốc có thể làm tăng nồng độ imatinib trong huyết tương: Những chất làm ức chế hoạt động của cytochrome P450 isoenzyme CYP3A4 (ví dụ ketoconazole, itraconazole, erythromycin, clarithromycin) có thể làm giảm sự chuyển hóa và làm tăng nồng độ imatinib. Có sự tăng đáng kể về sự tồn lưu imatinib (nồng độ cao nhất trong huyết tương (Cmax) trung bình của imatinib tăng 26% và diện tích dưới đường cong nồng độ (AUC) của imatinib tăng 40%) ở những người khỏe mạnh khi thuốc được dùng đồng thời với một liều đơn ketoconazole (một chất ức chế CYP3A4). Nên thận trọng khi dùng Glivec với các chất ức chế nhóm CYP3A4.
Những thuốc có thể bị thay đổi nồng độ trong huyết tương do Glivec:
Glivec làm tăng Cmax trung bình và AUC của simvastatin (cơ chất CYP3A4) lần lượt là gấp 2 lần và gấp 3,5 lần, cho thấy sự ức chế CYP3A4 bởi Glivec. Vì vậy, khuyến cáo thận trọng khi dùng Glivec với các cơ chất CYP3A4 có cửa sổ điều trị hẹp (ví dụ cyclosporin hoặc pimozide). Glivec có thể làm tăng nồng độ trong huyết tương của các thuốc được chuyển hóa bởi CYP3A4 khác (ví dụ triazolo-benzodiazepine, thuốc chẹn kênh canxi nhóm dihydropyridine, một số chất ức chế HMG-CoA reductase tức là các statin, v.v…).
Glivec cũng ức chế hoạt động CYP2C9 và CYP2C19 in vitro. Đã quan sát thấy thời gian prothrombin (PT) kéo dài sau khi dùng đồng thời với warfarin. Vì vậy khi dùng coumarin, cần theo dõi thời gian prothrombin một thời gian ngắn lúc khởi đầu và lúc kết thúc điều trị Glivec và khi thay đổi liều. Một cách khác là nên cân nhắc sử dụng heparin trọng lượng phân tử thấp.
In vitro, Glivec ức chế hoạt động của cytochrome P450 isoenzyme CYP2D6 ở nồng độ tương tự như nồng độ ảnh hưởng đến hoạt động của CYP3A4. Glivec 400 mg 2 lần/ngày có tác dụng ức chế yếu lên sự chuyển hóa metoprolol qua trung gian CYP2D6, với Cmax và AUC của metoprolol tăng khoảng 23%. Dùng đồng thời Glivec với các cơ chất CYP2D6 như metoprolol dường như không phải là yếu tố nguy cơ gây tương tác thuốc-thuốc và việc điều chỉnh liều có thể là không cần thiết.
In vitro, Glivec ức chế con đường chuyển hóa của acetaminophen O-glucuronidate (Ki 58,5microM).
Dùng đồng thời Glivec (400 mg/ngày trong 8 ngày) với acetaminophen/paracetamol (1000 mg liều duy nhất vào ngày thứ 8) ở bệnh nhân bị CML không dẫn đến bất kỳ thay đổi nào về mặt dược động học của acetaminophen/paracetamol.
Dược động học của Glivec không bị thay đổi khi dùng acetaminophen/paracetamol đơn liều.
Chưa có dữ liệu về độ an toàn và dược động học khi dùng đồng thời Glivec ở liều lớn hơn 400 mg/ngày hoặc sử dụng lâu dài với acetaminophen/paracetamol.
Tác dụng ngoại ý
Tóm tắt về độ an toàn
Đặc tính chung về an toàn của Glivec trong sử dụng lâm sàng trên người đã được mô tả rõ qua hơn 12 năm kinh nghiệm sử dụng Glivec. Trong quá trình phát triển lâm sàng, phần lớn bệnh nhân trải qua các phản ứng phụ tại một số thời điểm. Các phản ứng phụ của thuốc thường gặp nhất (>10%) được báo cáo là: giảm bạch cầu trung tính, giảm tiểu cầu, thiếu máu, đau đầu, rối loạn tiêu hóa, phù, tăng cân, buồn nôn, nôn, chuột rút, đau cơ xương, tiêu chảy, phát ban, mệt mỏi, và đau bụng. Các phản ứng từ nhẹ đến trung bình, và chỉ có 2 đến 5% bệnh nhân phải ngừng điều trị vĩnh viễn do các phản ứng liên quan đến thuốc.
Độ an toàn của Glivec ở bệnh nhân người lớn và trẻ em bị bệnh bạch cầu Ph+ là tương tự nhau.
Sự khác nhau về đặc tính an toàn giữa bệnh bạch cầu Ph+ và khối u đặc là tỉ lệ ức chế tủy xương cao hơn và mức độ nghiêm trọng hơn trong bệnh bạch cầu Ph+, và xuất huyết đường tiêu hóa và xuất huyết trong các khối u ở bệnh nhân GIST và hầu như chắc chắn là do các yếu tố liên quan đến bệnh. Ức chế tủy xương, các phản ứng phụ ở đường tiêu hóa, phù và phát ban thường gặp ở hai nhóm bệnh nhân này. Các tình trạng bệnh lý của đường tiêu hóa khác, như là tắc nghẽn đường tiêu hóa, thủng và loét, dường như xuất hiện chuyên biệt theo chỉ định. Các phản ứng phụ nổi bật khác đã được quan sát thấy sau khi dùng Glivec, và có thể có quan hệ nhân quả, bao gồm nhiễm độc gan, suy thận cấp, giảm phosphate huyết, phản ứng bất lợi nghiêm trọng về đường hô hấp, hội chứng ly giải khối u và chậm tăng trưởng ở trẻ em.
Có thể cần phải điều chỉnh liều lượng, tùy thuộc vào mức độ nghiêm trọng của phản ứng. Rất ít trường hợp phải ngừng thuốc do các phản ứng phụ của thuốc.
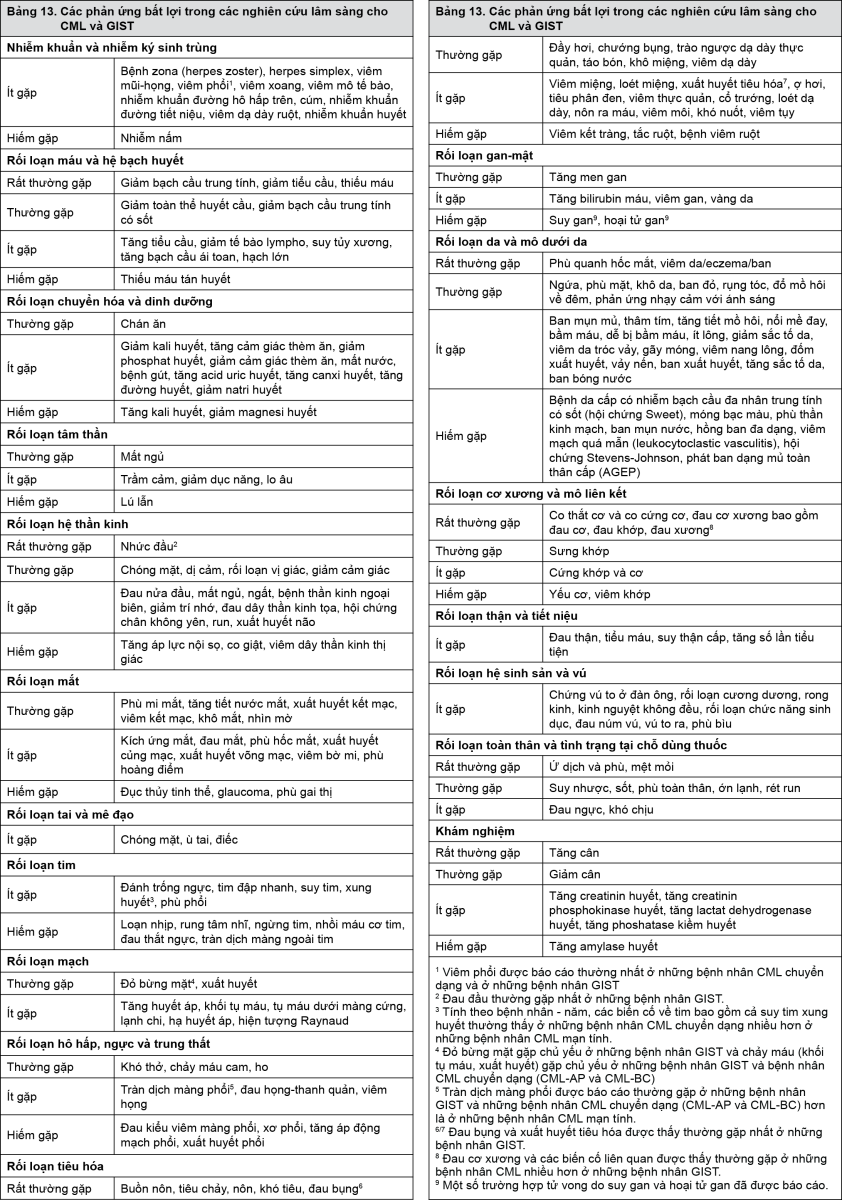
Các phản ứng bất lợi (bảng 13 và bảng 14) được liệt kê theo tiêu đề về tần suất, đầu tiên là hay gặp nhất, sử dụng quy ước sau đây: Rất thường gặp (≥1/10), thường gặp (≥1/100, <1/10), ít gặp (≥1/1000, <1/100), hiếm gặp (≥1/10.000, <1/1000), rất hiếm gặp (<1/10.000) kể cả các báo cáo riêng lẻ. Các phản ứng bất lợi và tần suất của chúng được báo cáo trong bảng 13 dựa trên các nghiên cứu đăng ký cho CML và GIST.
- xem Bảng 13.
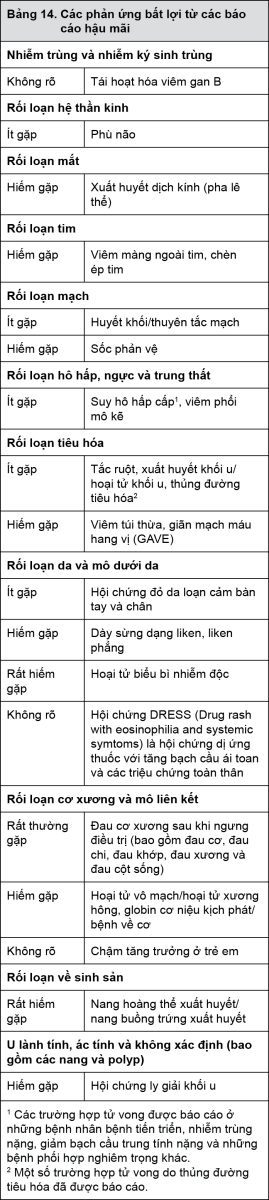
Các dạng phản ứng phụ của thuốc sau đây được báo cáo từ kinh nghiệm hậu mãi và từ các nghiên cứu lâm sàng thêm với Glivec. Các phản ứng phụ này bao gồm các báo cáo tự phát cũng như các phản ứng phụ nghiêm trọng của thuốc từ các nghiên cứu lâm sàng nhỏ hơn hoặc các nghiên cứu đang tiến hành và các chương trình tiếp cận mở rộng. Do các phản ứng của thuốc này được báo cáo từ một quần thể với dân số không rõ, nên không thể ước tính một cách đáng tin cậy tần suất của chúng hay thiết lập quan hệ nhân quả với việc sử dụng Glivec.
- xem Bảng 14.
Mô tả các phản ứng phụ chọn lọc của thuốc
Ức chế tủy xương
Ức chế tủy xương rất phổ biến ở bệnh nhân ung thư điều trị bằng Glivec. Ức chế tủy xương, giảm tiểu cầu, giảm bạch cầu trung tính, và thiếu máu là những bất thường độ 3 và 4 về xét nghiệm thường gặp nhất đã được báo cáo. Nhìn chung, sự ức chế tủy xương ở bệnh nhân bị CML dùng Glivec thường hồi phục và ở hầu hết bệnh nhân không gây gián đoạn liều lượng hoặc giảm liều. Một số ít bệnh nhân cần ngừng thuốc. Các phản ứng khác như giảm toàn bộ huyết cầu, giảm lympho bào và ức chế tủy xương cũng đã được báo cáo.
Giảm tế bào máu có thể xuất hiện nhiều nhất ở liều cao nhất và xuất hiện phụ thuộc vào giai đoạn của bệnh CML, giảm bạch cầu trung cầu trung tính và giảm tiểu cầu độ 3 hoặc 4 ở mức cao hơn gấp 4 đến 6 lần trong cơn nguyên bào và giai đoạn tăng tốc (44% đối với giảm bạch cầu trung tính và 63% đối với giảm tiểu cầu) khi so với bệnh nhân bị bệnh bạch cầu tủy mạn giai đoạn mạn tính mới được chẩn đoán (16,7% giảm bạch cầu trung tính và 8,9% giảm tiểu cầu). Những biến cố này thường có thể xử trí bằng cách giảm liều hoặc ngừng điều trị, nhưng hiếm khi cần ngừng điều trị bằng Glivec. Tỉ lệ độc tính huyết học ở những bệnh nhân có khối u đặc (ví dụ như GIST) thấp hơn các bệnh nhân bị bệnh bạch cầu với nhiễm sắc thể Philadelphia dương tính, khoảng 10% giảm bạch cầu trung tính độ 3 hoặc 4 và 1% giảm tiểu cầu độ 3 hoặc 4.
Xuất huyết
Xuất huyết hệ thần kinh trung ương và xuất huyết tiêu hóa không hiếm gặp ở bệnh nhân bị bệnh bạch cầu tủy mạn tính có chức năng tủy đã bị tổn thương từ lúc ban đầu. Xuất huyết là một dấu hiệu dễ nhận biết của biến chứng của bệnh trong nhóm đối tượng bị bệnh bạch cầu cấp tính, có thể là kết quả của giảm tiểu cầu, hoặc ít gặp hơn là rối loạn chức năng tiểu cầu. Tuy nhiên, không phải tất cả bệnh nhân bị xuất huyết hệ thần kinh trung ương và xuất huyết tiêu hóa trong quá trình điều trị bằng imatinib có giảm tiểu cầu.
Biểu hiện phổ biến nhất của chảy máu có ý nghĩa lâm sàng là xuất huyết tiêu hóa, thường xảy ra nhất ở các bệnh nhân bị bạch cầu tủy mạn tiến triển và ở các bệnh nhân GIST di căn, trong đó chảy máu có thể xảy ra như là một phần của bệnh chính do chảy máu khối u vì xuất huyết khối u/hoại tử khối u. Đã quan sát thấy tần suất xuất huyết tiêu hóa thường thấp nhất trong bối cảnh điều trị bước một CML và điều trị bổ trợ GIST. Cũng hiếm có báo cáo hậu mãi về giãn mạch máu hang vị (GAVE) khi điều trị bằng Glivec.
Phù và ứ dịch
Phù là một độc tính phổ biến của imatinib xuất hiện trên hơn 50% bệnh nhân của tất cả các chỉ định. Phù có liên quan đến liều và có mối tương quan giữa sự xuất hiện của phù và nồng độ thuốc trong huyết tương. Biểu hiện thường gặp nhất là phù quanh mắt và ít gặp hơn là phù chi dưới. Thường không cần phải điều trị đặc hiệu. Các biến cố ứ dịch khác ít gặp hơn, nhưng do ở vị trí giải phẫu nên có thể nghiêm trọng. Biến cố ứ dịch thường gặp nhất là tràn dịch màng phổi, thường gặp nhất ở các bệnh nhân bị bệnh bạch cầu tủy mạn tiến triển và GIST di căn. Tần suất bị suy tim thường thấp ở các bệnh nhân bị phù và ứ dịch. Tần suất này cao hơn ở bệnh nhân bị bệnh bạch cầu tủy mạn tiến triển so với các nhóm khác. Điều này được giải thích do tình trạng sức khỏe ở các bệnh nhân bị bệnh bạch cầu tủy mạn tiến triển kém hơn. Cũng nhận thấy xu hướng tương tự về suy thận ở các bệnh nhân bị phù và ứ dịch.
Trong một nghiên cứu lâm sàng, tần suất của các biến cố gợi ý có suy tim xung huyết là 1,5% ở nhóm imatinib so với 1,1% của nhóm dùng IFN-alpha ở bệnh nhân bị bạch cầu tủy mạn mới chẩn đoán. Tần suất cao hơn đáng kể ở các bệnh nhân bị bạch cầu tủy mạn chuyển dạng (giai đoạn tăng tốc hoặc cơn nguyên bào), tuổi cao hơn, hoặc với mức hemoglobin lúc ban đầu nhỏ hơn 8g/dL. Suy tim xung huyết và rối loạn chức năng thất trái được tiếp tục theo dõi trong các báo cáo cập nhật tính an toàn định kỳ (PSUR: periodic safety update report). Trong tất cả các chỉ định, nhận thấy bệnh nhân bệnh bạch cầu tủy mạn có tần suất suy tim xung huyết cao hơn so với bệnh nhân GIST, có thể chỉ ra sự khác nhau của một số yếu tố nguy cơ liên quan đến bệnh. Ngoài ra, một phân tích tính an toàn đặc biệt được công bố gần đây về các biến cố tim trong nghiên cứu EORTC trên 942 bệnh nhân bị GIST không thể cắt bỏ được hoặc GIST di căn đã kết luận rằng imatinib không dẫn đến suy tâm thất trái ở bệnh nhân GIST với tỉ lệ quan sát được khoảng 0,2% trong khi nó có thể lên tới 2% đối tượng đã bị bệnh tim.
Ban da và các phản ứng bất lợi về da nghiêm trọng
Đã có báo cáo về hồng ban toàn thân, dát sần, phát ban ngứa có thể biến mất dù vẫn tiếp tục điều trị. Một số bệnh nhân có thể ngứa mà không kèm theo ban, và đôi khi là một phần da bị tróc. Một số bệnh nhân xuất hiện lại ban da khi dùng thuốc trở lại, nhưng không phải ở tất cả các bệnh nhân. Những ban da này thường có đáp ứng với thuốc kháng histamin và thuốc steroid tại chỗ. Đôi khi cần sử dụng steroid toàn thân.
Đã quan sát thấy ban da ở 1/3 bệnh nhân được điều trị bằng imatinib cho tất cả các chỉ định. Thường xảy ra ngứa và thường gặp nhất là sang thương hồng ban, dát sần hoặc tróc da trên cánh tay, thân hoặc mặt hay biểu hiện trên toàn thân. Sinh thiết da cho thấy một phản ứng với độc tính thuốc với thâm nhiễm tế bào hỗn hợp. Mặc dù hầu hết các phát ban là nhẹ và tự giới hạn, những trường hợp nặng hiếm gặp như hoại tử biểu bì nhiễm độc Stevens-Johnson, hồng ban đa dạng hoặc DRESS có thể cần phải gián đoạn hoặc ngừng điều trị.
Không ngạc nhiên khi các phản ứng da được quan sát với một tỷ lệ cao hơn so với giả dược trong thử nghiệm điều trị bổ trợ GIST.
Nhiễm độc gan: Nhiễm độc gan, đôi khi nghiêm trọng có thể xảy ra đã quan sát thấy tiền lâm sàng và lâm sàng. Các bất thường chức năng gan thường bao gồm tăng nhẹ transaminase, mặc dù một số ít bệnh nhân tăng mức bilirubin. Đợt tấn công thường trong vòng hai tháng điều trị đầu tiên, nhưng cũng có xảy ra muộn sau 6 đến 12 tháng sau khi bắt đầu điều trị. Nồng độ trở về mức bình thường sau khi ngừng điều trị 1 đến 4 tuần.
Giảm phosphat huyết
Phosphat huyết thanh thấp và giảm phosphat huyết (lên đến độ 3 hoặc 4) đã được quan sát thấy tương đối phổ biến ở tất cả các chỉ định, tuy nhiên không chứng minh được nguồn gốc và ý nghĩa lâm sàng của phát hiện này. Imatinib đã có biểu hiện ức chế sự biệt hóa bạch cầu đơn nhân thành hủy cốt bào. Sự suy giảm này đi kèm giảm khả năng tiêu hủy xương ở các tế bào này. Đã quan sát thấy sự giảm phụ thuộc liều của RANK-L trong các hủy cốt bào với sự có mặt của imatinib. Sự ức chế luôn được duy trì của hoạt động của các hủy cốt bào có thể dẫn tới đáp ứng điều hòa ngược, kết quả là tăng nồng độ của PTH. Sự liên quan lâm sàng của những phát hiện tiền lâm sàng chưa rõ ràng và sự kết hợp với các phản ứng bất lợi trên xương như gãy xương chưa được chứng minh.
Trong các chương trình phát triển lâm sàng, phosphat huyết thanh không được đo thường qui trong tất cả các nghiên cứu. Mặc dù bước đầu đã có giả thuyết rằng giảm phosphat huyết có thể phụ thuộc liều, nhưng kết quả 24 tháng phân tích được từ nghiên cứu pha III TOPS được thiết kế để khảo sát sự phụ thuộc liều của các kết cục về an toàn trên bệnh nhân CML mới được chẩn đoán đã cho thấy rằng phosphat huyết thanh giảm độ 3 hoặc 4 là 19,1% so với 15,5% và calci huyết thanh giảm độ 3 hoặc 4 là 5,1% so với 0,9% lần lượt ở bệnh nhân dùng 400 mg và 800 mg.
Tắc, thủng hoặc loét đường tiêu hóa: Loét đường tiêu hóa, có thể được coi là đại diện cho các trường hợp kích ứng tại chỗ quá mức do dùng imatinib, đã được quan sát thấy trên một tỷ lệ nhỏ bệnh nhân ở tất cả các chỉ định. Xuất huyết khối u/hoại tử khối u, tắc và thủng đường tiêu hóa được xem như bệnh liên quan và xảy ra duy nhất hoặc thường xuyên hơn ở các bệnh nhân GIST. Trong trường hợp GIST di căn, hoại tử khối u có thể xảy ra như là phản ứng của khối u, hiếm khi dẫn đến thủng. Tắc đường tiêu hóa/liệt ruột xảy ra phổ biến nhất ở các bệnh nhân GIST, khi đó nguyên nhân có thể là do khối u GIST di căn gây tắc nghẽn và trong trường hợp điều trị hỗ trợ do dính ruột từ phẫu thuật đường tiêu hóa trước kia.
Hội chứng ly giải khối u: Có thể có mối quan hệ nhân quả giữa hội chứng ly giải khối u và điều trị bằng Glivec, mặc dù một số trường hợp bị gây nhiễu bởi các thuốc dùng đồng thời và các nguy cơ không phụ thuộc khác (xem phần Cảnh báo).
Chậm tăng trưởng ở trẻ em: Glivec có vẻ ảnh hưởng đến tầm vóc của trẻ, đặc biệt ở trẻ em trước tuổi dậy thì. Không thể loại trừ mối quan hệ nhân quả giữa việc chậm tăng trưởng ở trẻ và điều trị bằng Glivec, mặc dù thông tin chỉ giới hạn ở một số trường hợp chậm tăng trưởng trên bệnh nhân CML (xem phần Cảnh báo).
Phản ứng phụ của thuốc về đường hô hấp nặng: Phản ứng nặng về đường hô hấp, đôi khi gây tử vong, đã được quan sát với điều trị bằng Glivec, bao gồm suy hô hấp cấp tính, tăng huyết áp động mạch phổi, bệnh phổi kẽ và xơ phổi. Các bệnh lý tim hoặc phổi có sẵn có thể liên quan đến các biến cố hô hấp nặng đã được báo cáo trong nhiều trường hợp trên.
Các bất thường về xét nghiệm
Huyết học
Giảm tế bào máu do CML, đặc biệt là giảm bạch cầu trung tính và giảm tiểu cầu là một dấu hiệu luôn gặp trong tất cả các nghiên cứu, với tần suất cao hơn ở những liều cao ≥ 750mg (nghiên cứu giai đoạn I). Tuy nhiên, sự xuất hiện giảm tế bào máu cũng phụ thuộc rõ vào giai đoạn của bệnh. Ở những bệnh nhân bị bệnh bạch cầu tủy mạn mới được chẩn đoán, giảm tế bào máu ít gặp hơn so với các bệnh nhân bị bệnh bạch cầu tủy mạn khác. Tần suất giảm bạch cầu trung tính ở độ 3 hoặc 4 (ANC < 1,0x109/L) và giảm tiểu cầu (số lượng tiểu cầu < 50x109/L) ở mức cao hơn gấp 4-6 lần trong cơn nguyên bào và giai đoạn tăng tốc (59-64% đối với giảm bạch cầu trung tính và 44-63% đối với giảm tiểu cầu) khi so với bệnh nhân bị bệnh bạch cầu tủy mạn giai đoạn mạn tính mới được chẩn đoán (16,7% giảm bạch cầu trung tính và 8,9% giảm tiểu cầu). Ở bệnh nhân bị bệnh bạch cầu tủy mạn giai đoạn mãn tính mới được chẩn đoán, đã quan sát thấy giảm bạch cầu trung tính độ 4 (ANC < 0,5x109/L) trong số 3,6% bệnh nhân và giảm tiểu cầu độ 4 (số lượng tiểu cầu < 10x109/L) < 1% bệnh nhân. Thời gian trung vị của các giai đoạn giảm bạch cầu trung tính thường trong khoảng từ 2 đến 3 tuần và giảm tiểu cầu thường trong khoảng 3 đến 4 tuần. Những trường hợp này thường có thể xử trí bằng cách giảm liều hoặc gián đoạn điều trị bằng Glivec, nhưng trong những trường hợp hiếm gặp có thể dẫn đến ngừng điều trị hoàn toàn. Ở bệnh nhi bị bệnh bạch cầu tủy mạn, độc tính thường gặp nhất là giảm tế bào máu độ 3 hoặc 4 bao gồm giảm bạch cầu trung tính, giảm tiểu cầu và thiếu máu. Những phản ứng này thường xảy ra trong vài tháng đầu điều trị.
Ở bệnh nhân bị GIST ác tính không thể cắt bỏ hoặc đã di căn (nghiên cứu B2222), thiếu máu độ 3 đã được báo cáo ở 5,4% bệnh nhân và thiếu máu độ 4 ở 0,7% bệnh nhân và có thể liên quan với chảy máu dạ dày ruột hoặc chảy máu bên trong khối u ít nhất ở một số trong những bệnh nhân này. Đã gặp giảm bạch cầu trung tính độ 3 ở 7,5% và độ 4 ở 2,7% bệnh nhân, và giảm tiểu cầu độ 3 ở 0,7% bệnh nhân. Không có bệnh nhân nào giảm tiểu cầu ở độ 4. Giảm bạch cầu và bạch cầu trung tính chủ yếu xảy ra trong 6 tuần đầu điều trị, sau đó các trị số giữ tương đối ổn định.
Sinh hóa
Đã ghi nhận tăng nghiêm trọng các transaminase (< 5%) hoặc bilirubin (< 1%) ở bệnh nhân bị bệnh bạch cầu tủy mạn (CML) và thường được xử trí bằng cách giảm liều hoặc ngừng thuốc (thời gian trung vị của giai đoạn này khoảng 1 tuần). Ngừng điều trị hoàn toàn do các xét nghiệm gan bất thường là dưới 1% bệnh nhân CML. Ở bệnh nhân bị u mô đệm dạ dày ruột (GIST) (nghiên cứu B2222), đã quan sát thấy tăng SGPT (glutamic pyruvic transferase huyết thanh) độ 3 hoặc độ 4 là 6,8% và tăng SGOT (glutamic oxaloacetic transferase huyết thanh) độ 3 hoặc độ 4 là 4,8%. Tăng bilirubin dưới 3%.
Đã có các trường hợp viêm gan hủy tế bào, viêm gan ứ mật và suy gan, trong đó một số trường hợp kết cục là tử vong.
Bảo quản
Không bảo quản ở nhiệt độ trên 30oC và tránh ẩm. Giữ thuốc trong bao bì gốc.
Phân loại ATC
L01XE01 - imatinib
Trình bày/Đóng gói
Viên nén bao phim: hộp 6 vỉ x 10 viên.
- Abacavir
- Abernil
- Abiiogran
- Acarbose
- ACC
- Acebutolol
- Acenocoumarol
- Acetate Ringer's
- Acetazolamide
- Acetylcystein
- Acetylsalicylic acid
- Aciclovir
- Acid acetylsalicylic
- Acid aminocaproic
- Acid ascorbic
- Acid boric
- Acid chenodeoxycholic
- Acid ethacrynic
- Acid folic
- Acid fusidic
- Acid iopanoic
- Acid ioxaglic
- Acid nalidixic
- Acid pantothenic
- Acid para-aminobenzoic
- Acid salicylic
- Acid tranexamic
- Acid valproic
- Acid zoledronic
- Acitretin
- Aclasta
- Aclon
- Actapulgite
- Actelsar
- Actelsar HCT
- Actemra
- Actilyse
- Acular
- Acupan
- Acuvail
- Acyclovir STADA
- Acyclovir STADA Cream
- Adalat
- Adenosin
- Adenosin Ebewe
- Adipiodon
- Advagraf
- Aerius
- Afinitor
- Agicarvir
- Agifovir-E
- Agilosart
- Agilosart-H
- Agimepzol
- Agimosarid
- Agimstan
- Agimstan-H
- Agiremid
- Agivastar
- Aibezym
- Air-X
- Alaxan
- Albendazol
- Albiomin
- Albumin
- Albumin người Grifols 20%
- Albuminar
- AlbuRx
- Albutein
- Alcuronium chloride
- Aldesleukin
- Alendronat
- Alertin
- Alfa-Lipogamma 600 Oral
- Alfuzosin hydrochlorid
- Algotra
- Alimemazin
- Alimta
- Allipem
- Allopurinol
- Allopurinol STADA
- Aloxi
- Alprazolam
- Alpha Chymotrypsin
- Alpha tocopherol
- Alphachymotrypsin Glomed
- Alphagan-P
- Aluvia
- Alzental
- Amaryl
- Ambroco
- Ambroxol
- Amcinol-Paste
- Amigold
- Amikacin
- Aminocaproic acid
- Aminoleban
- Aminoleban Oral
- Aminosteril N-Hepa
- Amiparen
- Amitriptyline
- Amiyu
- Amlodipine
- Amlor
- Amoxicillin
- Amoxicillin & clavulanate
- Ampicillin
- Amquitaz
- Anaferon for children
- Anargil
- Anaropin
- Andriol Testocaps
- Anepzil
- Anyfen
- Apaisac
- Apidra SoloStar
- Apitim 5
- Aprovel
- Aquaphil
- Arcalion
- Arcoxia
- Aricept Evess
- Arimidex
- Arnetine
- Artrodar
- A-Scabs
- Ascorbic acid
- Asperlican/Candinazol
- Aspilets EC
- Aspirin
- Asthmatin
- Atelec
- Atocib 120
- Atocib 90
- Atosiban PharmIdea
- Atozet
- Attapulgite
- Atussin
- Atropin
- Augbactam
- Augmentin Sachet
- Augmentin SR
- Augmentin Tablets
- Augmex
- Avamys
- Avastin
- Avelox Dịch truyền
- Avelox Viên nén
- Avodart
- Axcel Cefaclor-125 Suspension
- Axcel Cetirizine Syrup
- Axcel Chlorpheniramine
- Axcel Dexchlorpheniramine
- Axcel Dicyclomine-S Syrup
- Axcel Diphenhydramine Paediatric Syrup
- Axcel Erythromycin ES
- Axcel Eviline
- Axcel Fungicort Cream
- Axcel Fusidic acid Cream
- Axcel Fusidic acid-B Cream
- Axcel Hydrocortisone Cream
- Axcel Lignocaine 2% Sterile Gel
- Axcel Loratadine
- Axcel Miconazole Cream
- Axcel Paracetamol
- Axcel Urea Cream
- Axitan
- Azenmarol
- Azicine
- Aziphar
- Azithromycin
Quảng cáo




